

Proteogenomics produces comprehensive and highly accurate protein-coding gene annotation in a complete genome assembly of Malassezia sympodialis
- PMID: 28100699
- PMCID: PMC5389616
- DOI: 10.1093/nar/gkx006
Proteogenomics produces comprehensive and highly accurate protein-coding gene annotation in a complete genome assembly of Malassezia sympodialis
Abstract
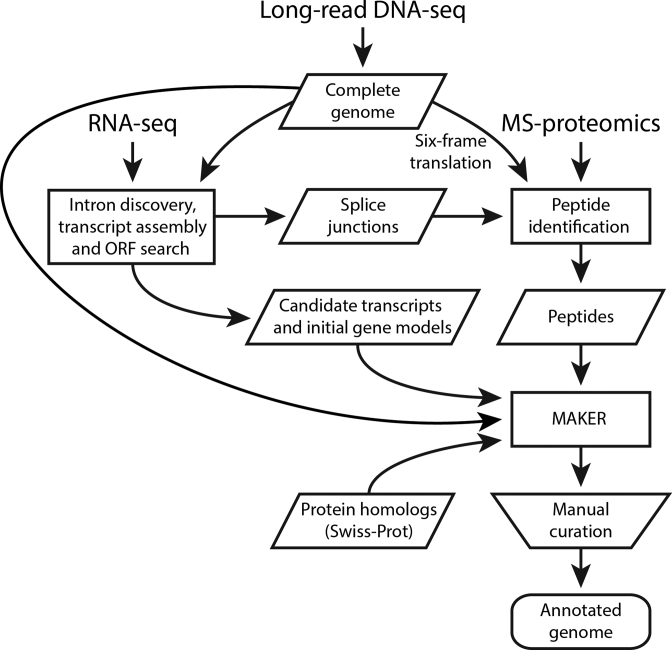
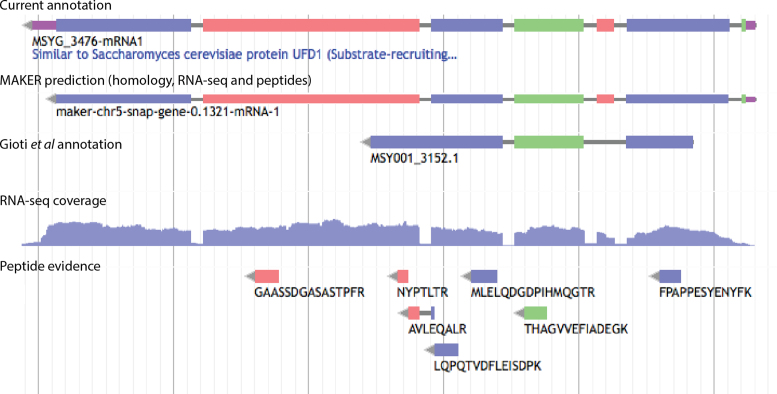
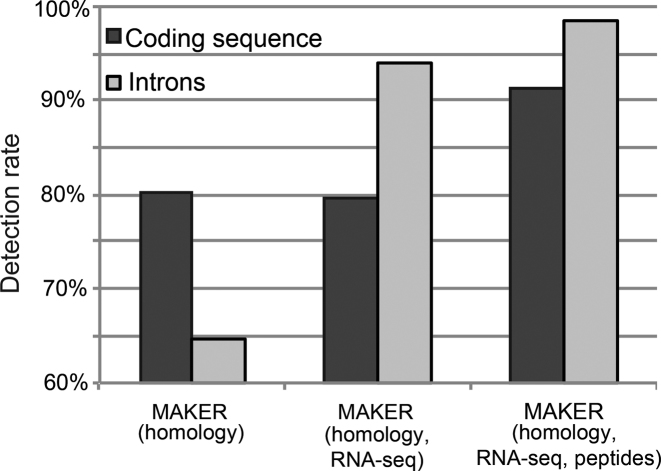
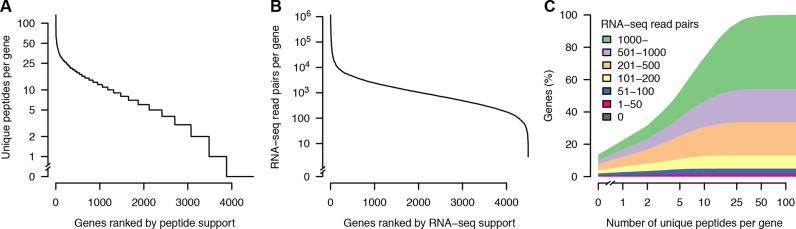
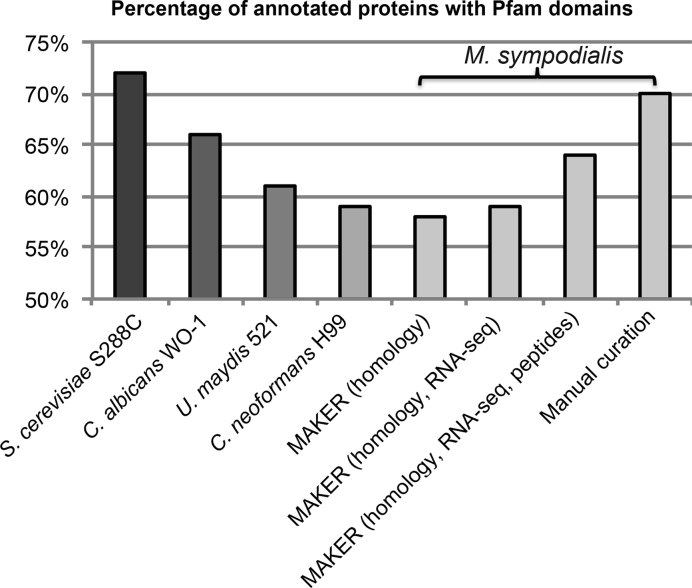
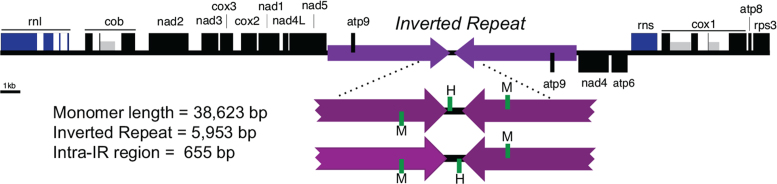
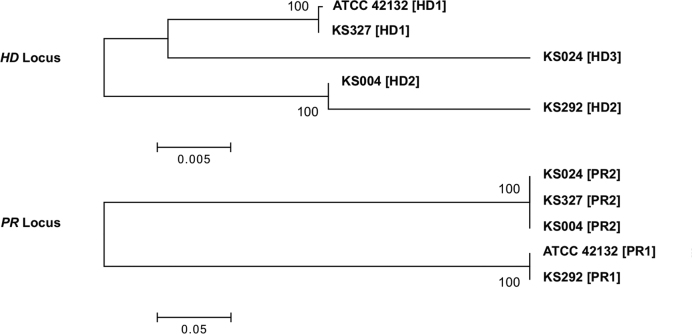
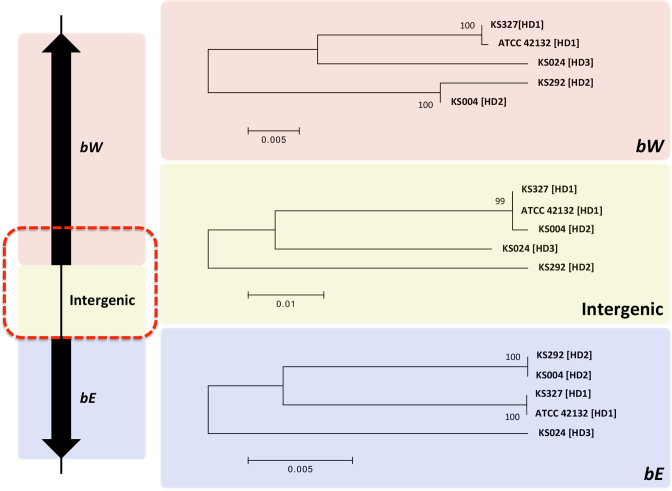
Complete and accurate genome assembly and annotation is a crucial foundation for comparative and functional genomics. Despite this, few complete eukaryotic genomes are available, and genome annotation remains a major challenge. Here, we present a complete genome assembly of the skin commensal yeast Malassezia sympodialis and demonstrate how proteogenomics can substantially improve gene annotation. Through long-read DNA sequencing, we obtained a gap-free genome assembly for M. sympodialis (ATCC 42132), comprising eight nuclear and one mitochondrial chromosome. We also sequenced and assembled four M. sympodialis clinical isolates, and showed their value for understanding Malassezia reproduction by confirming four alternative allele combinations at the two mating-type loci. Importantly, we demonstrated how proteomics data could be readily integrated with transcriptomics data in standard annotation tools. This increased the number of annotated protein-coding genes by 14% (from 3612 to 4113), compared to using transcriptomics evidence alone. Manual curation further increased the number of protein-coding genes by 9% (to 4493). All of these genes have RNA-seq evidence and 87% were confirmed by proteomics. The M. sympodialis genome assembly and annotation presented here is at a quality yet achieved only for a few eukaryotic organisms, and constitutes an important reference for future host-microbe interaction studies.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
