

Differential Vulnerability of CA1 versus CA3 Pyramidal Neurons After Ischemia: Possible Relationship to Sources of Zn2+ Accumulation and Its Entry into and Prolonged Effects on Mitochondria
- PMID: 28100752
- PMCID: PMC5242414
- DOI: 10.1523/JNEUROSCI.3270-16.2016
Differential Vulnerability of CA1 versus CA3 Pyramidal Neurons After Ischemia: Possible Relationship to Sources of Zn2+ Accumulation and Its Entry into and Prolonged Effects on Mitochondria
Abstract
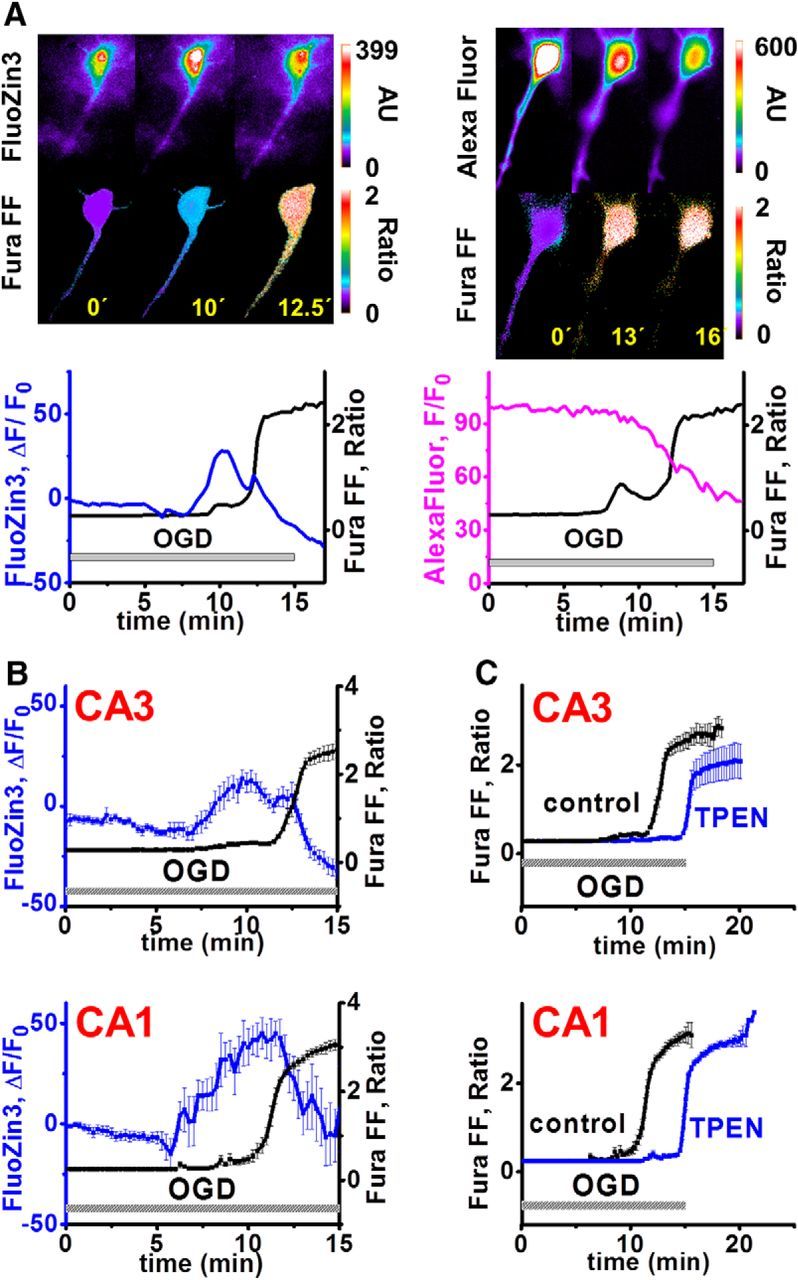
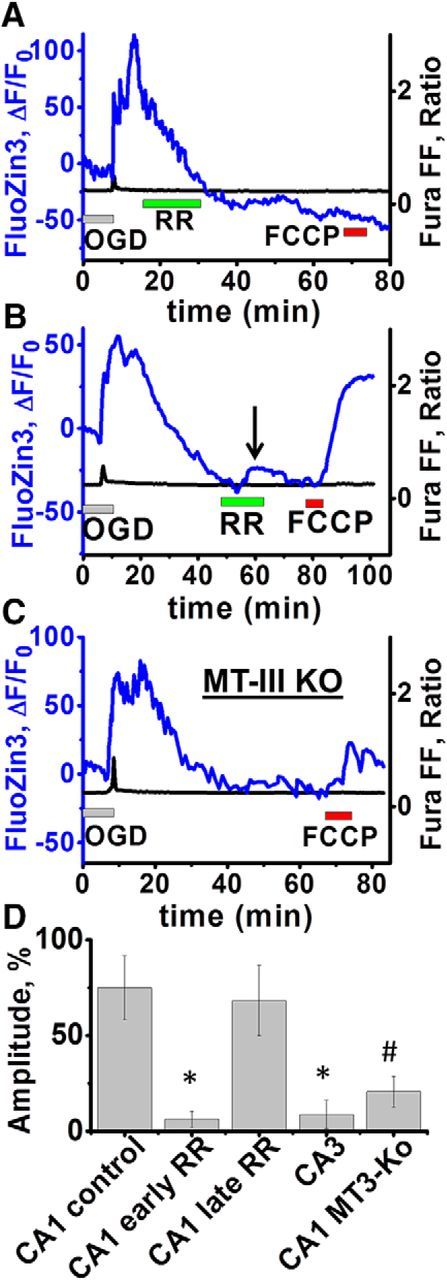
Excitotoxic mechanisms contribute to the degeneration of hippocampal pyramidal neurons after recurrent seizures and brain ischemia. However, susceptibility differs, with CA1 neurons degenerating preferentially after global ischemia and CA3 neurons after limbic seizures. Whereas most studies address contributions of excitotoxic Ca2+ entry, it is apparent that Zn2+ also contributes, reflecting accumulation in neurons either after synaptic release and entry through postsynaptic channels or upon mobilization from intracellular Zn2+-binding proteins such as metallothionein-III (MT-III). Using mouse hippocampal slices to study acute oxygen glucose deprivation (OGD)-triggered neurodegeneration, we found evidence for early contributions of excitotoxic Ca2+ and Zn2+ accumulation in both CA1 and CA3, as indicated by the ability of Zn2+ chelators or Ca2+ entry blockers to delay pyramidal neuronal death in both regions. However, using knock-out animals (of MT-III and vesicular Zn2+ transporter, ZnT3) and channel blockers revealed substantial differences in relevant Zn2+ sources, with critical contributions of presynaptic release and its permeation through Ca2+- (and Zn2+)-permeable AMPA channels in CA3 and Zn2+ mobilization from MT-III predominating in CA1. To assess the consequences of the intracellular Zn2+ accumulation, we used OGD exposures slightly shorter than those causing acute neuronal death; under these conditions, cytosolic Zn2+ rises persisted for 10-30 min after OGD, followed by recovery over ∼40-60 min. Furthermore, the recovery appeared to be accompanied by mitochondrial Zn2+ accumulation (via the mitochondrial Ca2+ uniporter MCU) in CA1 but not in CA3 neurons and was markedly diminished in MT-III knock-outs, suggesting that it depended upon Zn2+ mobilization from this protein.
Significance statement: The basis for the differential vulnerabilities of CA1 versus CA3 pyramidal neurons is unclear. The present study of events during and after acute oxygen glucose deprivation highlights a possible important difference, with rapid synaptic entry of Ca2+ and Zn2+ contributing more in CA3, but with delayed and long-lasting accumulation of Zn2+ within mitochondria occurring in CA1 but not CA3 pyramidal neurons. These data may be consistent with observations of prominent mitochondrial dysfunction as a critical early event in the delayed degeneration of CA1 neurons after ischemia and support a hypothesis that mitochondrial Zn2+ accumulation in the early reperfusion period may be a critical and targetable upstream event in the injury cascade.
Keywords: CA1 pyramidal neurons; delayed degeneration; hippocampal slice; in vitro ischemia model; mitochondria; oxygen glucose deprivation.
Copyright © 2017 the authors 0270-6474/17/370726-12$15.00/0.
Figures






References
-
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem. 2000;75:1878–1888. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous