

Genomic Infectious Disease Epidemiology in Partially Sampled and Ongoing Outbreaks
- PMID: 28100788
- PMCID: PMC5850352
- DOI: 10.1093/molbev/msw275
Genomic Infectious Disease Epidemiology in Partially Sampled and Ongoing Outbreaks
Abstract
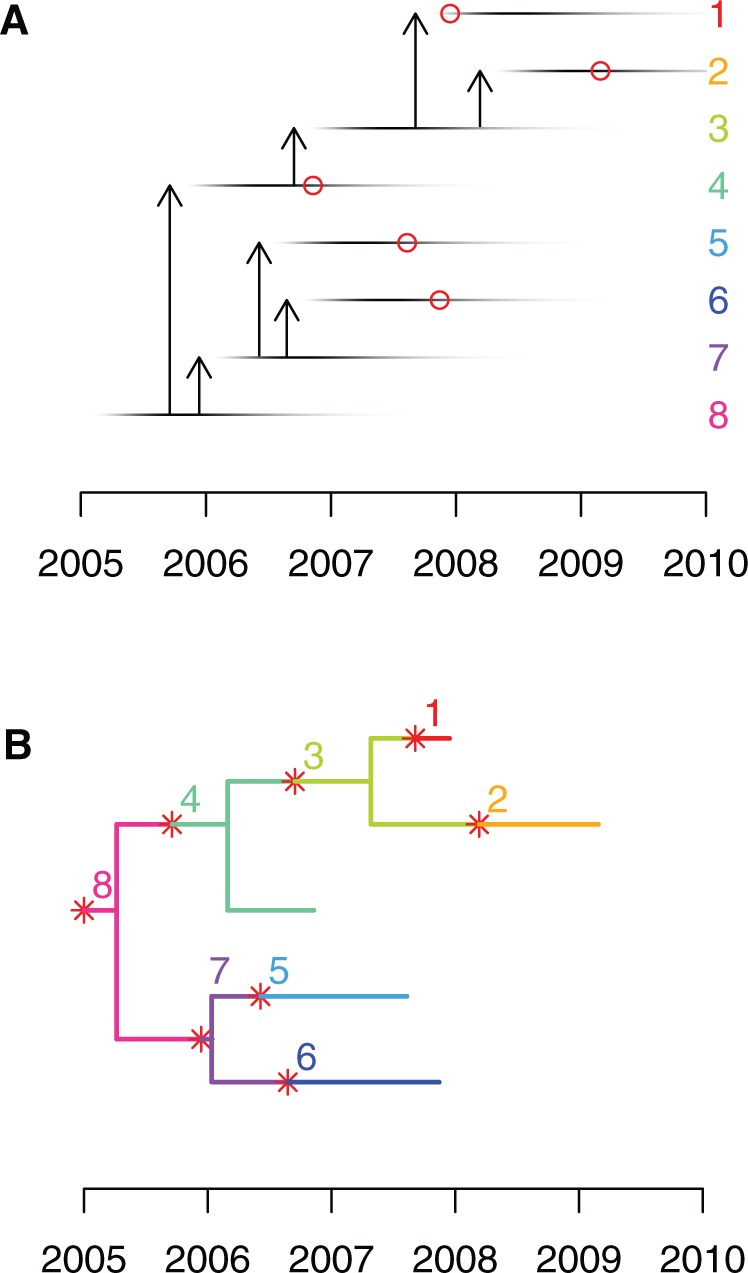
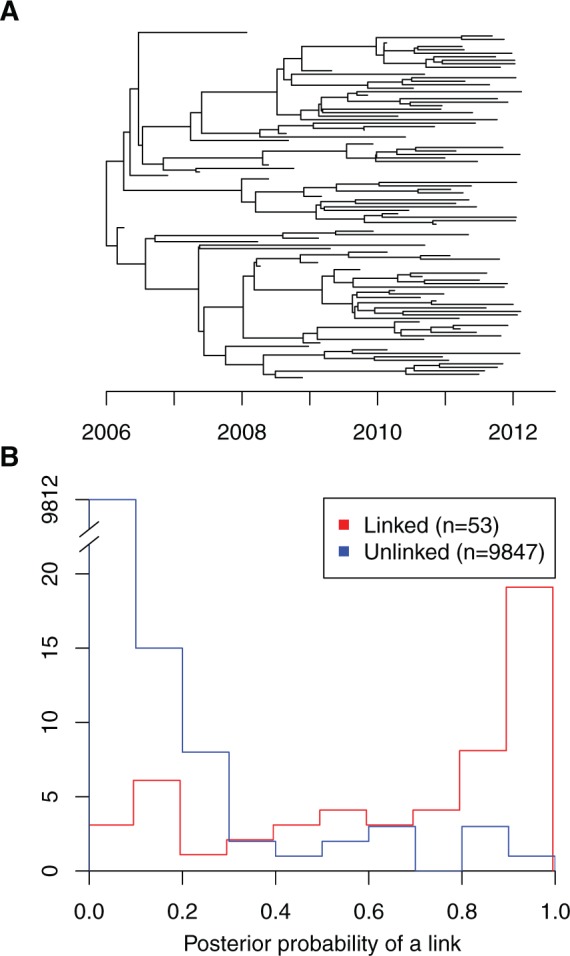
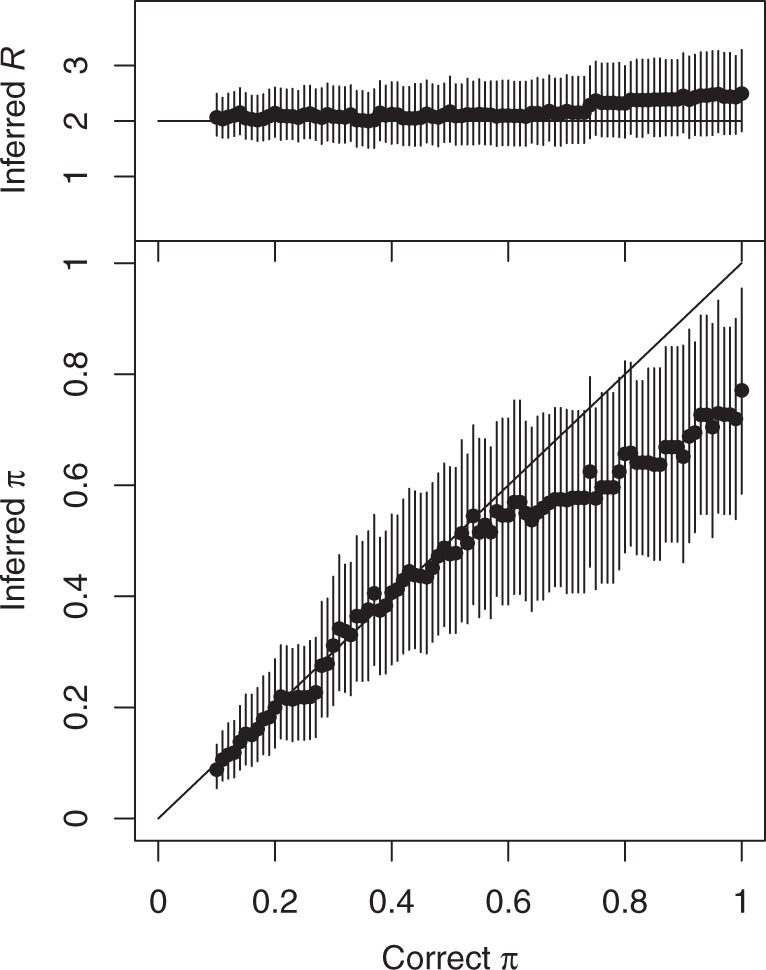
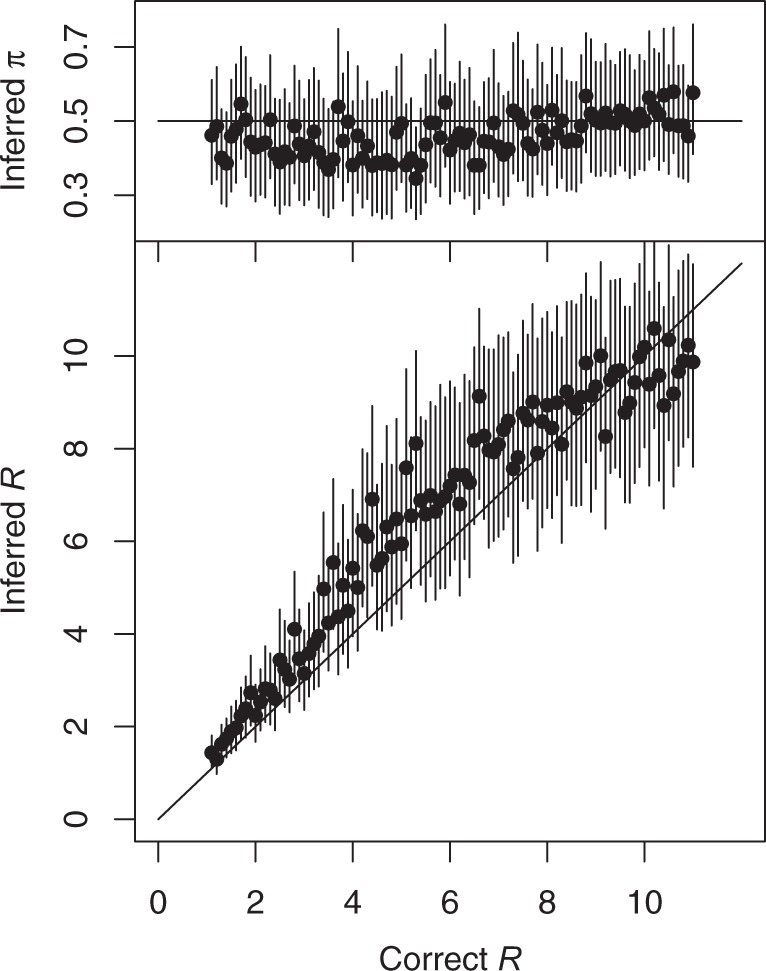
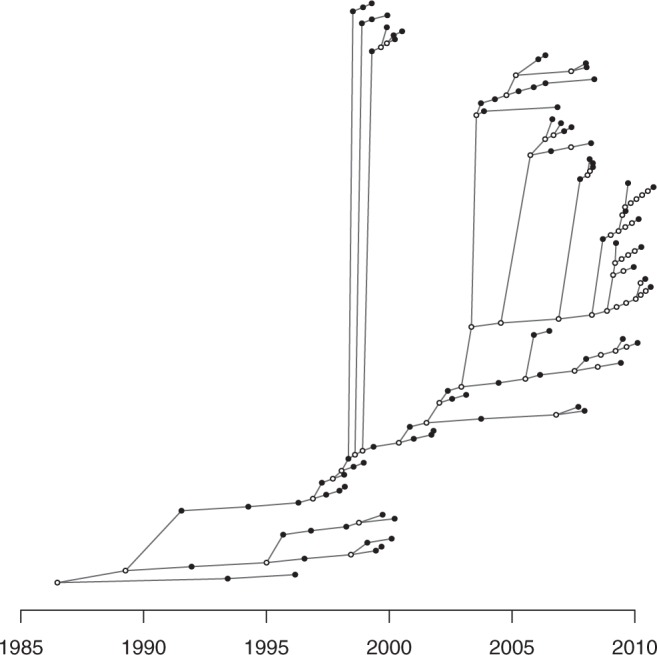
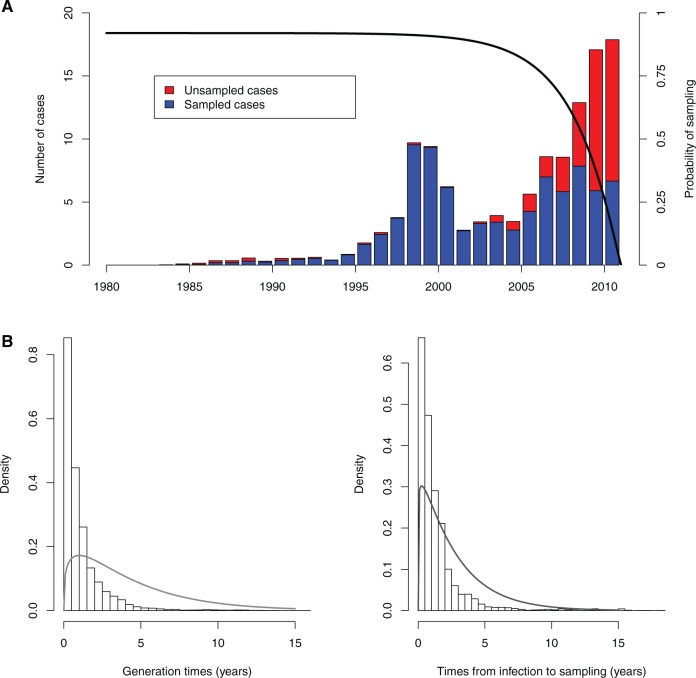
Genomic data are increasingly being used to understand infectious disease epidemiology. Isolates from a given outbreak are sequenced, and the patterns of shared variation are used to infer which isolates within the outbreak are most closely related to each other. Unfortunately, the phylogenetic trees typically used to represent this variation are not directly informative about who infected whom-a phylogenetic tree is not a transmission tree. However, a transmission tree can be inferred from a phylogeny while accounting for within-host genetic diversity by coloring the branches of a phylogeny according to which host those branches were in. Here we extend this approach and show that it can be applied to partially sampled and ongoing outbreaks. This requires computing the correct probability of an observed transmission tree and we herein demonstrate how to do this for a large class of epidemiological models. We also demonstrate how the branch coloring approach can incorporate a variable number of unique colors to represent unsampled intermediates in transmission chains. The resulting algorithm is a reversible jump Monte-Carlo Markov Chain, which we apply to both simulated data and real data from an outbreak of tuberculosis. By accounting for unsampled cases and an outbreak which may not have reached its end, our method is uniquely suited to use in a public health environment during real-time outbreak investigations. We implemented this transmission tree inference methodology in an R package called TransPhylo, which is freely available from https://github.com/xavierdidelot/TransPhylo.
Keywords: genomic epidemiology; infectious disease outbreak; transmission analysis.
© The Author 2017. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Anderson RM, May RM.. 1992. Infectious diseases of humans: dynamics and control. Oxford: Oxford University Press.
-
- Azarian T, Daum RS, Petty LA, Steinbeck JL, Yin Z, Nolan D, Boyle-Vavra S, Hanage WP, Salemi M, David MZ, et al. 2016. Intrahost evolution of Methicillin-resistant Staphylococcus aureus USA300 among individuals with reoccurring skin and soft-tissue infections. J Infect Dis. 214:895–905. - PMC - PubMed
-
- Becker N. 1977. Estimation for discrete time branching processes with application to epidemics. Biometrics 33:515–522. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
