

Blind prediction of deleterious amino acid variations with SNPs&GO
- PMID: 28102005
- PMCID: PMC5522651
- DOI: 10.1002/humu.23179
Blind prediction of deleterious amino acid variations with SNPs&GO
Abstract
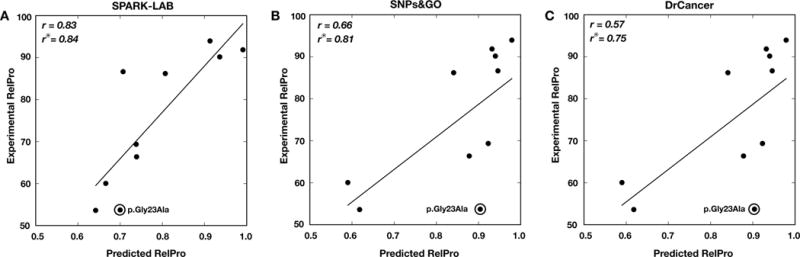
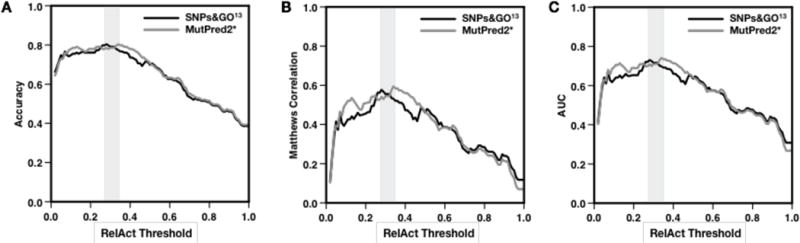
SNPs&GO is a machine learning method for predicting the association of single amino acid variations (SAVs) to disease, considering protein functional annotation. The method is a binary classifier that implements a support vector machine algorithm to discriminate between disease-related and neutral SAVs. SNPs&GO combines information from protein sequence with functional annotation encoded by gene ontology (GO) terms. Tested in sequence mode on more than 38,000 SAVs from the SwissVar dataset, our method reached 81% overall accuracy and an area under the receiving operating characteristic curve of 0.88 with low false-positive rate. In almost all the editions of the Critical Assessment of Genome Interpretation (CAGI) experiments, SNPs&GO ranked among the most accurate algorithms for predicting the effect of SAVs. In this paper, we summarize the best results obtained by SNPs&GO on disease-related variations of four CAGI challenges relative to the following genes: CHEK2 (CAGI 2010), RAD50 (CAGI 2011), p16-INK (CAGI 2013), and NAGLU (CAGI 2016). Result evaluation provides insights about the accuracy of our algorithm and the relevance of GO terms in annotating the effect of the variants. It also helps to define good practices for the detection of deleterious SAVs.
Keywords: disease-related variation; gene ontology; genome interpretation; machine learning; protein function; single amino acid variation; variant annotation.
© 2017 Wiley Periodicals, Inc.
Conflict of interest statement
The authors declare that they have no conflict of interests.
Figures
References
-
- Brownstein CA, Beggs AH, Homer N, Merriman B, Yu TW, Flannery KC, DeChene ET, Towne MC, Savage SK, Price EN, et al. An international effort towards developing standards for best practices in analysis, interpretation and reporting of clinical genome sequencing results in the CLARITY Challenge. Genome Biol. 2014;15(3):R53. - PMC - PubMed
-
- Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum Mutat. 2009;30(8):1237–44. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
