

Scaling single-cell genomics from phenomenology to mechanism
- PMID: 28102262
- PMCID: PMC5438464
- DOI: 10.1038/nature21350
Scaling single-cell genomics from phenomenology to mechanism
Abstract
Three of the most fundamental questions in biology are how individual cells differentiate to form tissues, how tissues function in a coordinated and flexible fashion and which gene regulatory mechanisms support these processes. Single-cell genomics is opening up new ways to tackle these questions by combining the comprehensive nature of genomics with the microscopic resolution that is required to describe complex multicellular systems. Initial single-cell genomic studies provided a remarkably rich phenomenology of heterogeneous cellular states, but transforming observational studies into models of dynamics and causal mechanisms in tissues poses fresh challenges and requires stronger integration of theoretical, computational and experimental frameworks.
Figures
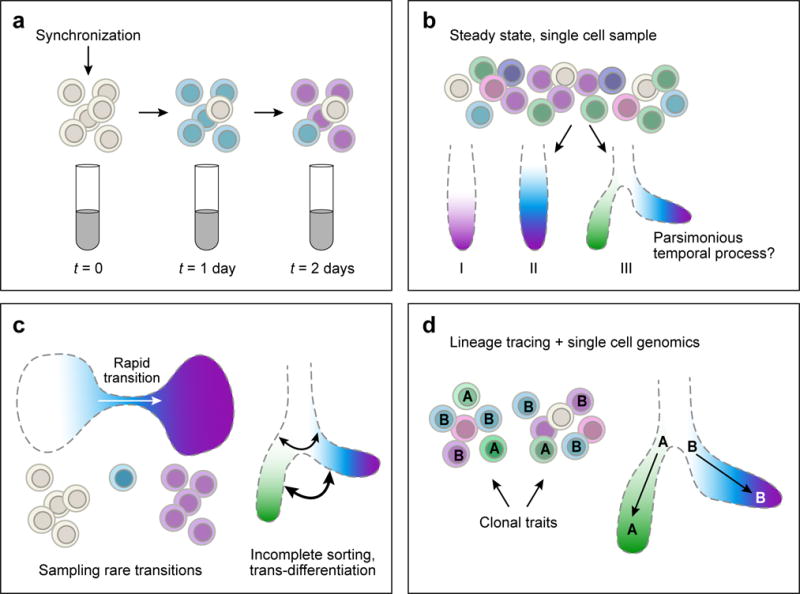
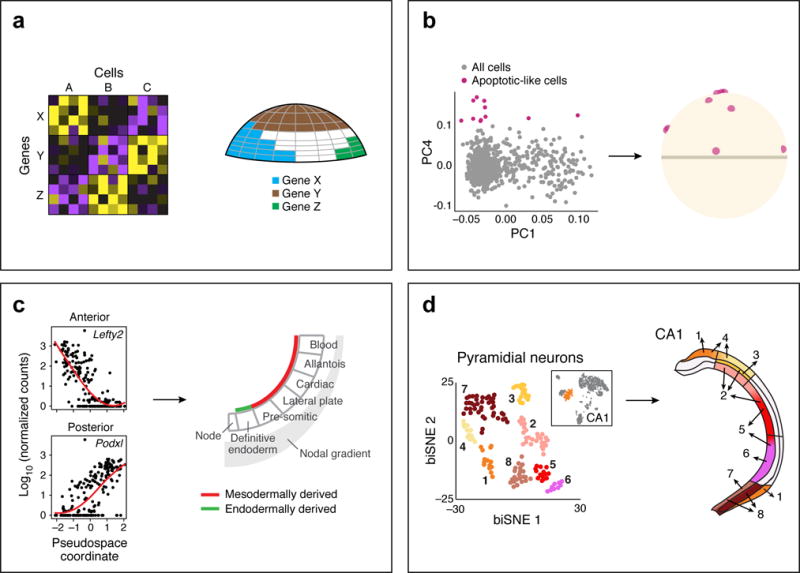
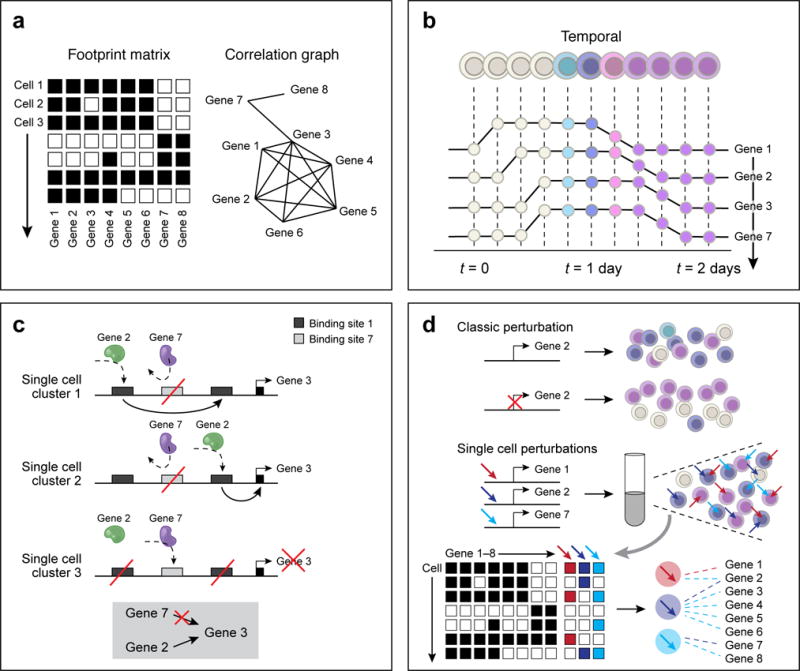
References
-
- Mayr E. The Growth of Biological Thought: Diversity, Evolution, and Inheritance. Belknap Press; 1982.
-
- Szathmary E, Maynard-Smith J. The Major Transitions in Evolution. Oxford; Univeristy Press: 1995.
-
- Gould SJ. Ontogeny and Phylogeny. Belknap Press; 1977.
