

Ataluren and similar compounds (specific therapies for premature termination codon class I mutations) for cystic fibrosis
- PMID: 28102546
- PMCID: PMC6464785
- DOI: 10.1002/14651858.CD012040.pub2
Ataluren and similar compounds (specific therapies for premature termination codon class I mutations) for cystic fibrosis
Update in
-
Ataluren and similar compounds (specific therapies for premature termination codon class I mutations) for cystic fibrosis.Cochrane Database Syst Rev. 2023 Mar 3;3(3):CD012040. doi: 10.1002/14651858.CD012040.pub3. Cochrane Database Syst Rev. 2023. PMID: 36866921 Free PMC article.
Abstract
Background: Cystic fibrosis is a common life-shortening genetic disorder in the Caucasian population (less common in other ethnic groups) caused by the mutation of a single gene that codes for the production of the cystic fibrosis transmembrane conductance regulator protein. This protein coordinates the transport of salt (and bicarbonate) across cell surfaces and the mutation most notably affects the airways. In the lungs of people with cystic fibrosis, defective protein results in a dehydrated surface liquid and compromised mucociliary clearance. The resulting thick mucus makes the airway prone to chronic infection and inflammation, which consequently damages the structure of the airways, eventually leading to respiratory failure. Additionally, abnormalities in the cystic fibrosis transmembrane conductance regulator protein lead to other systemic complications including malnutrition, diabetes and subfertility.Five classes of mutation have been described, depending on the impact of the mutation on the processing of the cystic fibrosis transmembrane conductance regulator protein in the cell. In class I mutations, the presence of premature termination codons prevents the production of any functional protein resulting in a severe cystic fibrosis phenotype. Advances in the understanding of the molecular genetics of cystic fibrosis has led to the development of novel mutation-specific therapies. Therapies targeting class I mutations (premature termination codons) aim to mask the abnormal gene sequence and enable the normal cellular mechanism to read through the mutation, potentially restoring the production of the cystic fibrosis transmembrane conductance regulator protein. This could in turn make salt transport in the cells function more normally and may decrease the chronic infection and inflammation that characterises lung disease in people with cystic fibrosis.
Objectives: To evaluate the benefits and harms of ataluren and similar compounds on clinically important outcomes in people with cystic fibrosis with class I mutations (premature termination codons).
Search methods: We searched the Cochrane Cystic Fibrosis Trials Register which is compiled from electronic database searches and handsearching of journals and conference abstract books. We also searched the reference lists of relevant articles. Last search of Group's register: 24 October 2016.We searched clinical trial registries maintained by the European Medicines Agency, the US National Institutes of Health and the WHO. Last search of clinical trials registries: 28 November 2016.
Selection criteria: Randomised controlled trials of parallel design comparing ataluren and similar compounds (specific therapies for class I mutations) with placebo in people with cystic fibrosis who have at least one class I mutation. Cross-over trials were reviewed individually to evaluate whether data from the first treatment arm could be included. We excluded trials that combined therapies for premature termination codon class I mutations with other mutation-specific therapies.
Data collection and analysis: The authors independently assessed the risk of bias and extracted data from the included trial; they contacted trial authors for additional data.
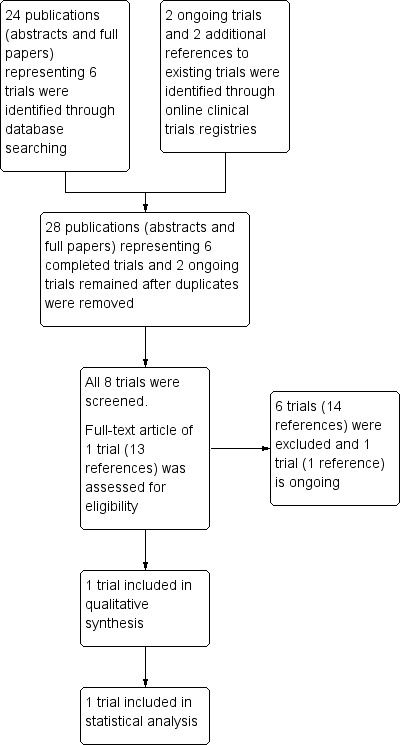
Main results: Our searches identified 28 references to eight trials; five trials were excluded (three were cross-over and one was not randomised and one did not have relevant outcomes), one cross-over trial is awaiting classification pending provision of data and one trial is ongoing. The included parallel randomised controlled trial compared ataluren to placebo for a duration of 48 weeks in 238 participants (age range 6 to 53 years) with cystic fibrosis who had at least one nonsense mutation (a type of class I mutation).The quality of evidence and risk of bias assessments for the trial were moderate overall. Random sequence generation, allocation concealment and blinding of trial personnel were well-documented; participant blinding was less clear. Some participant data were excluded from the analysis. The trial was assessed as high risk of bias for selective outcome reporting, especially when reporting on the trial's post hoc subgroup of participants by chronic inhaled antibiotic use.The trial was sponsored by PTC Therapeutics Incorporated with grant support by the Cystic Fibrosis Foundation, the Food and Drug Administration's Office of Orphan Products Development and the National Institutes of Health (NIH).The trial reported no significant difference between treatment groups in quality of life, assessed by the Cystic Fibrosis Questionnaire-Revised respiratory domain score and no improvement in respiratory function measures (mean difference of relative change in forced expiratory volume at one second 2.97% (95% confidence interval -0.58 to 6.52)). Ataluren was associated with a significantly higher rate of episodes of renal impairment, risk ratio 17.70 (99% confidence interval 1.28 to 244.40). The trial reported no significant treatment effect for ataluren for the review's secondary outcomes: pulmonary exacerbation; computerised tomography score; weight; body mass index; and sweat chloride. No deaths were reported in the trial.A post hoc subgroup analysis of participants not receiving chronic inhaled tobramycin (n = 146) demonstrated favourable results for ataluren (n = 72) for relative change in % predicted forced expiratory volume at one second and pulmonary exacerbation rate. Participants receiving chronic inhaled tobramycin appeared to have a reduced rate of pulmonary exacerbation compared to those not receiving chronic inhaled tobramycin. This drug interaction was not anticipated and may affect the interpretation of the trial results.
Authors' conclusions: There is currently insufficient evidence to determine the effect of ataluren as a therapy for people with cystic fibrosis with class I mutations. Future trials should carefully assess for adverse events, notably renal impairment and consider the possibility of drug interactions. Cross-over trials should be avoided given the potential for the treatment to change the natural history of cystic fibrosis.
Conflict of interest statement
There are no declarations of interest for any of the authors.
Figures

References
References to studies included in this review
Kerem 2014 {published data only}
-
- Ajayi T, Konstan M, Accurso FJ, Boeck K, Kerem E, Rowe S, et al. The use of high resolution computerized tomography of the chest in evaluating the effect of ataluren in nonsense mutation cystic fibrosis (nmCF) lung disease [abstract]. Journal of Cystic Fibrosis 2013;12 Suppl 1:S64, Abstract no: 63. [CENTRAL: 921666; CFGD Register: BD167h; CRS: 5500100000011670]
-
- Davies JC, Tiddens HAWM, Malfroot A, Heijerman HGM, Kerem E, Hjelte L, Sun J, et al. Ataluren in nonsense mutation cystic fibrosis patients not receiving tobramycin: significant lung function benefits in the paediatric age range. Journal of Cystic Fibrosis : Official Journal of the European Cystic Fibrosis Society 2016;15 Suppl 1:S21, Abstractt no: WS13.11. [CENTRAL: 1171482; CFGD Register: BD167q; CRS: 5500135000001597]
-
- Boeck K, Heijerman HGM, Davies JC, Sermet‐Gaudelus I, Hjelte L, Kerem E, et al. Ataluren significantly reduces exacerbations in nonsense mutation cystic fibrosis patients not receiving tobramycin. Journal of Cystic Fibrosis : Official Journal of the European Cystic Fibrosis Society 2016;15 Suppl 1:S20, Abstract no: WS13.1. [CENTRAL: 1171483; CFGD Register: BD167p; CRS: 5500135000001598]
-
- DeBoeck K, Sermet‐Gaudelus I, Kerem E, Wilschanski M, Accurso FJ, Konstan M, et al. Design of the ataluren confirmatory phase 3, randomised, double‐blind, placebo‐controlled trial in patients with nonsense mutation cystic fibrosis (ACT CF) [abstract]. Pediatric Pulmonology 2014;49 Suppl 38:312, Abstract no: 269. [CENTRAL: 1012387; CFGD Register: BD167n; CRS: 5500131000000160]
-
- Kerem E, Konstan MW, Boeck K, Accurso FJ, Sermet‐Gaudelus I, Wilschanski M, et al. Ataluren for the treatment of nonsense‐mutation cystic fibrosis: A randomised, double‐blind, placebo‐controlled phase 3 trial. The Lancet. Respiratory Medicine 2014;2(7):539‐47. [CENTRAL: 994980; CFGD Register: BD167l; CRS: 5500050000000189; EMBASE: 2014458185] - PMC - PubMed
References to studies excluded from this review
Kerem 2008 {published data only}
-
- Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet 2008;372:719‐27. - PubMed
-
- Kerem E, Yaakov Y, Armoni S, Pugatsch T, Shoseyov D, Cohen M, et al. PTC124 induces time‐dependent improvements in chloride conductance and clinical parameters in patients with nonsense‐mutation‐mediated cystic fibrosis [abstract]. Pediatric Pulmonology 2008;43 Suppl 31:294. [CENTRAL: 691199; CFGD Register: BD203b; CRS: 5500100000003330]
-
- Wilschanski M, Armoni S, Yaakov Y, Blau H, Shoseyov D, Cohen M, et al. PTC124 treatment over 3 months improves pharmacodynamic and clinical parameters in patients with nonsense ‐mutation‐mediated CF [abstract]. Journal of Cystic Fibrosis 2008;7(Suppl 2):S22. [CENTRAL: 651985; CFGD Register: BD203a; CRS: 5500100000003259]
-
- Wilschanski M, Miller LL, Shoseyov D, Blau H, Rivlin J, Aviram M, et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. European Respiratory Journal 2011;38:59‐69. - PubMed
McIntosh 2014b {published data only}
-
- McIntosh J. An open‐label safety and efficacy study for patients with nonsense mutation cystic fibrosis previously treated with ataluren (PTC124®). https://clinicaltrials.gov/ct2/show/NCT02107859 (accessed 23 February 2016). [clinicaltrials.gov: NCT02107859]
Pradal 2002 {published data only}
-
- Pradal U, Casotti V, Delmarco A, Nicolis E, Livraghi A, Conese M, et al. Effects of gentamicin on ion transport, MRNA and protein CFTR expression in patients with R1162X: A double blind placebo controlled study [abstract]. Pediatric Pulmonology 2002;Suppl 24:263. [CENTRAL: 404061; CFGD Register: BD145; CRS: 5500100000002232]
Romano 2000 {published data only}
-
- Romano L, Casciaro R, Vanini P, Zegarra‐Moran O, Negro I, Minuto N, et al. Reduction of sweat ion concentrations following topical application of gentamicin in CF patients carrying nonsense mutations [abstract]. 24th European Cystic Fibrosis Conference; 2001 June 6‐9; Vienna, Austria. 2001:P11. [CENTRAL: 354451; CFGD Register: BD144b; CRS: 5500100000001958]
-
- Romano L, Sacchi R, Zegarra‐Moran O, Vanini P, Guerriero F, Casciaro R, et al. Effects of topical applications of gentamicin on sweat test in CF patients carrying nonsense‐mutations [abstract]. Pediatric Pulmonology 2000;Suppl 20:250. [CENTRAL: 315387; CFGD Register: BD144a; CRS: 5500100000001755]
Wilschanski 2003 {published data only}
-
- Wilschanski M, Virgilis D, Strauss‐Liviatan N, Tal A, Bentur L, Blau H, et al. Gentamicin causes functional expression of CFTR in CF patients carrying stop mutations: a double‐blind placebo controlled trial [abstract]. Pediatric Pulmonology 2000;Suppl 20:244. [CENTRAL: 315400; CFGD Register: BD143a; CRS: 5500100000001763]
-
- Wilschanski M, Yahav J, Blau H, Bentur L, Rivlin J, Aviram M, et al. Restoration of CFTR function by gentamicin in cystic fibrosis patients carrying stop mutations: a double blind placebo controlled trial [abstract]. Gastroenterology 2003;124(4 Suppl 1):A582. [CENTRAL: 643127; CFGD Register: BD143c; CRS: 5500100000003235]
-
- Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, et al. Gentamicin‐induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. New England Journal of Medicine 2003;349(15):1433‐41. [CENTRAL: 440623; CFGD Register: BD143b; CRS: 5500100000002329; PUBMED: 14534336] - PubMed
References to studies awaiting assessment
Sermet‐Gaudelus 2010 {published data only}
-
- Sermet‐Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, Mogenet A, et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 2010;182(10):1262‐72. [CENTRAL: 768705; CFGD Register: BD22c; CRS: 5500100000006561] - PubMed
-
- Sermet‐Gaudelus I, Boeck K, Casimir G, Leal T, Vermeulen F, Mogenet A, et al. Children with nonsense‐mutation‐mediated cystic fibrosis respond to investigational treatment with PTC124 [abstract]. Pediatric Pulmonology 2008;43 Suppl 31:313. [CENTRAL: 689497; CFGD Register: BD22b; CRS: 5500100000003317]
-
- Sermet‐Gaudelus I, Leal T, Boeck K, Casimir G, Hanssens L, Hage P, et al. PTC124 induces CFTR full‐length production and activity in children with nonsense‐mutation‐mediated CF [abstract]. Journal of Cystic Fibrosis 2008;7(Suppl 2):S22. [CENTRAL: 651984; CFGD Register: BD22a; CRS: 5500100000003258]
References to ongoing studies
McIntosh 2014a {published data only}
-
- McIntosh J. A phase 3 efficacy and safety study of ataluren (PTC124®) in patients with nonsense mutation cystic fibrosis. https://clinicaltrials.gov/ct2/show/NCT02139306?term=NCT02139306&rank=1 (accessed 23 February 2016). [clinicaltrials.gov: NCT02139306]
Additional references
Boucher 2007
-
- Boucher RC. Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends in Molecular Medicine 2007;13(6):231–40. - PubMed
CFMD 2011
-
- Cystic Fibrosis Centre at the Hospital for Sick Children in Toronto, Canada. Cystic Fibrosis Mutation Database. www.genet.sickkids.on.ca/StatisticsPage.html (accessed 15 March 2015).
Clancy 2001
-
- Clancy JP, Bebök Z, Ruiz F, King C, Jones J, Walker L, et al. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 2001;163(7):1683‐92. - PubMed
Davis 2006
-
- Davis PB. Cystic fibrosis since 1938. American Journal of Respiratory and Critical Care Medicine 2006;173(5):475‐82. - PubMed
Deeks 2011
-
- Deeks J, Higgins J, Altman D on behalf of the Cochrane Statistical Methods Group (editors). Chapter 9 Analysing data and undertaking meta‐analysis. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Friedman 2014
-
- Friedman Y. Building Biotechnology. 4th Edition. Washington DC: Logo Press, 2014.
Fuchs 1994
-
- Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, et al. Effect of Aerosolized Recombinant Human DNase on Exacerbations of Respiratory Symptoms and on Pulmonary Function in Patients with Cystic Fibrosis. The New England Journal of Medicine 1994;331:637‐642. [DOI: 10.1056/NEJM199409083311003] - DOI - PubMed
Higgins 2003
Higgins 2011a
-
- Higgins JPT, Altman DG, Sterne JAC on behalf of the Cochrane Statistical Methods Group and the Cochrane Bias Methods Group (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
-
- Higgins JPT, Deeks JJ, Altman DG on behalf of the Cochrane Statistical Methods Group (editors). Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors). Cochrane Handbook of Systematic Reviews of Interventions. Version 5.1 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Howard 1996
-
- Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nature Medicine 1996;2(4):467‐9. - PubMed
Kerem 1997
-
- Kerem B, Chiba‐Falek O, Kerem E. Cystic fibrosis in Jews: frequency and mutation distribution. Genetic Testing 1997;1(1):35‐9. - PubMed
McElroy 2013
McKone 2006
-
- McKone EF, Goss CH, Aitken ML. CFTR genotype as a predictor of prognosis in cystic fibrosis. Chest 2006;130(5):1441‐47. - PubMed
Nicholson 2010
Patel 2015
Quittner 2009
-
- Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire‐Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest 2009;135(6):1610‐8. [DOI: 10.1378/chest.08-1190] - DOI - PMC - PubMed
Sinha 2014
UK CF Trust 2014
-
- CF Trust UK. Cystic fibrosis our focus. UK cystic fibrosis registry annual data report 2013. www.cysticfibrosis.org.uk/media/598466/annual‐data‐report‐2013‐jul14.pdf (accessed 15 March 2015).
Welch 2007
-
- Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007;447(7140):87‐91. - PubMed
Welsh 2001
-
- Welsh MJ, Ramsey BW, Accurso F, Cutting G. Cystic fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D editor(s). The Metabolic and Molecular Basis of Inherited Diseases. 8th Edition. New York: McGraw‐Hill, 2001:5121–88.
Wilschanski 2003
-
- Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, et al. Gentamicin induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. New England Journal of Medicine 2003;349(15):1433‐41. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical