

Copper-Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity
- PMID: 28103018
- PMCID: PMC5963733
- DOI: 10.1021/acs.chemrev.6b00636
Copper-Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity
Abstract
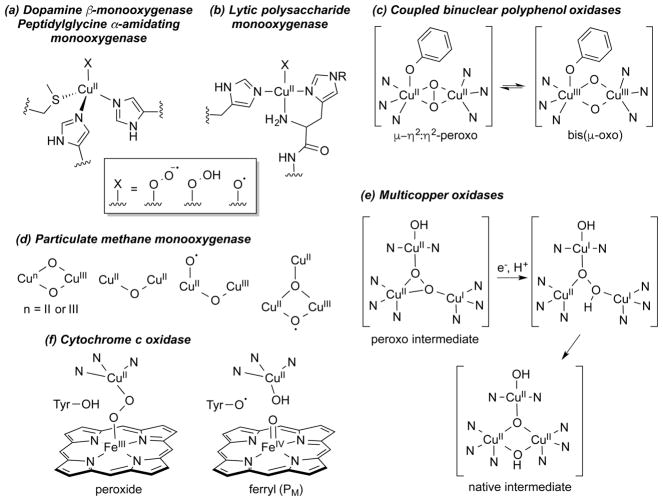
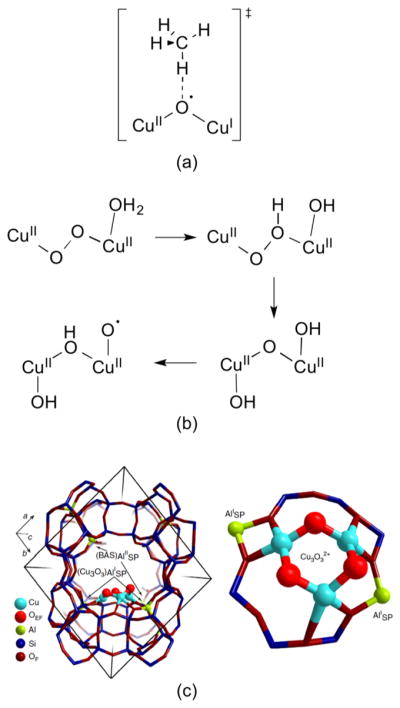
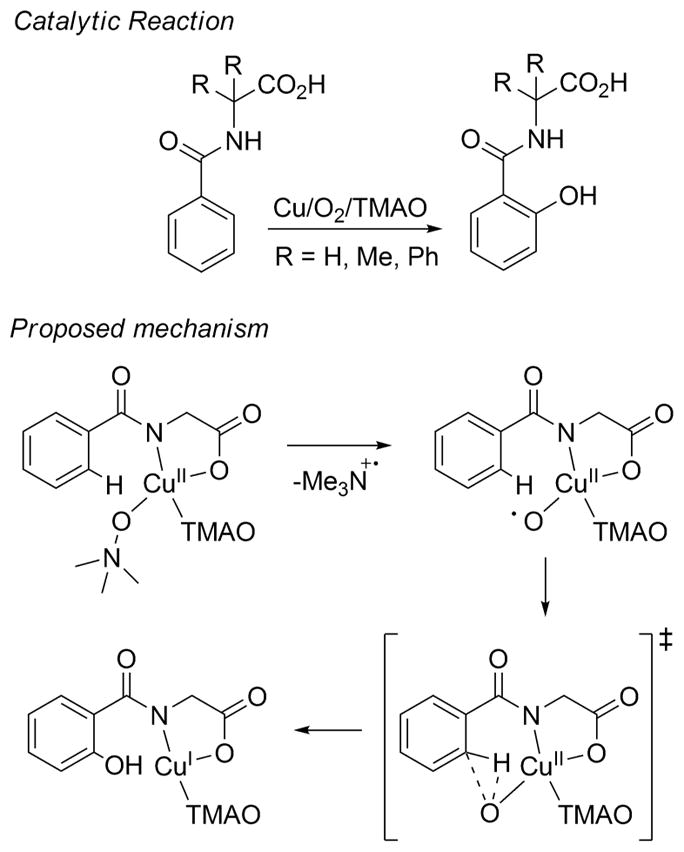
A longstanding research goal has been to understand the nature and role of copper-oxygen intermediates within copper-containing enzymes and abiological catalysts. Synthetic chemistry has played a pivotal role in highlighting the viability of proposed intermediates and expanding the library of known copper-oxygen cores. In addition to the number of new complexes that have been synthesized since the previous reviews on this topic in this journal (Mirica, L. M.; Ottenwaelder, X.; Stack, T. D. P. Chem. Rev. 2004, 104, 1013-1046 and Lewis, E. A.; Tolman, W. B. Chem. Rev. 2004, 104, 1047-1076), the field has seen significant expansion in the (1) range of cores synthesized and characterized, (2) amount of mechanistic work performed, particularly in the area of organic substrate oxidation, and (3) use of computational methods for both the corroboration and prediction of proposed intermediates. The scope of this review has been limited to well-characterized examples of copper-oxygen species but seeks to provide a thorough picture of the spectroscopic characteristics and reactivity trends of the copper-oxygen cores discussed.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
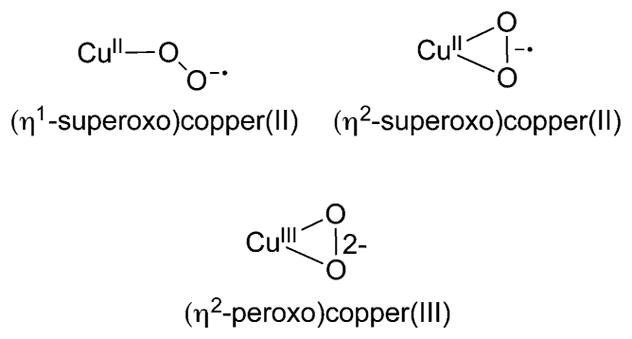
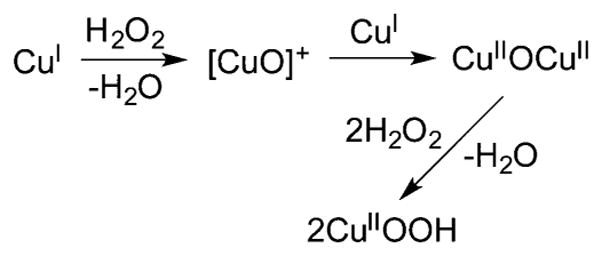
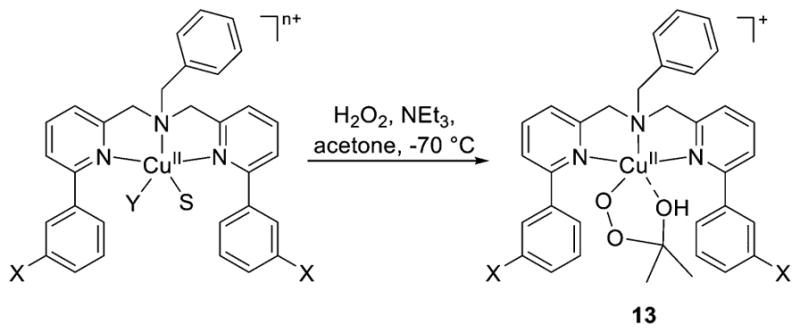
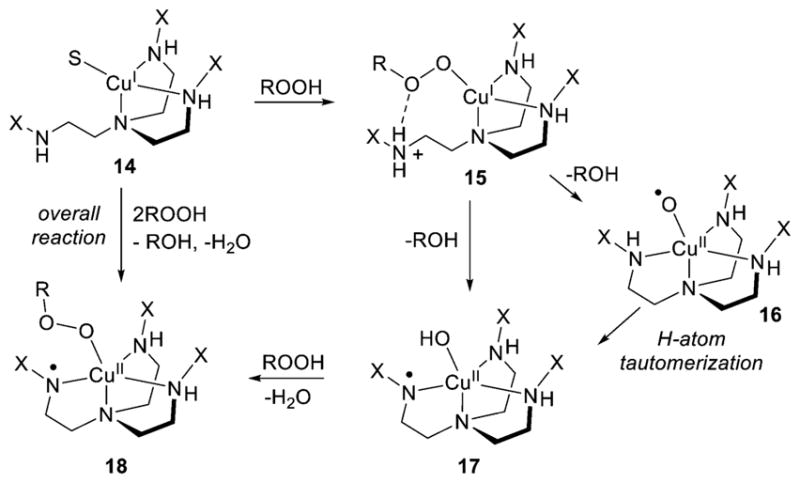
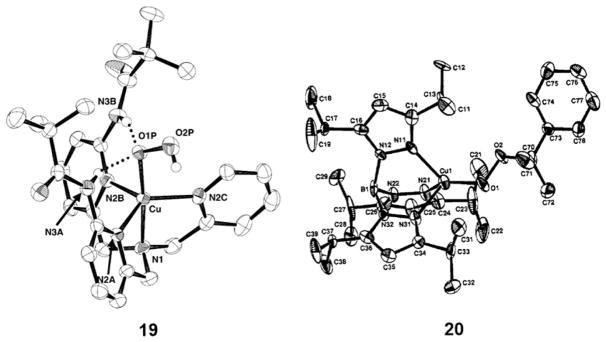
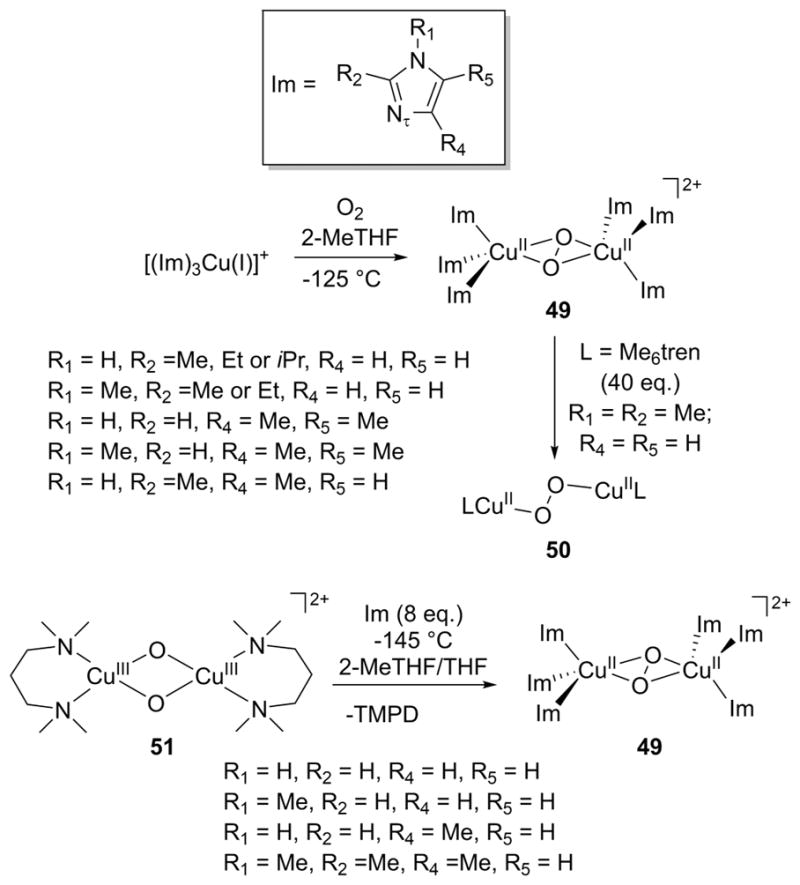
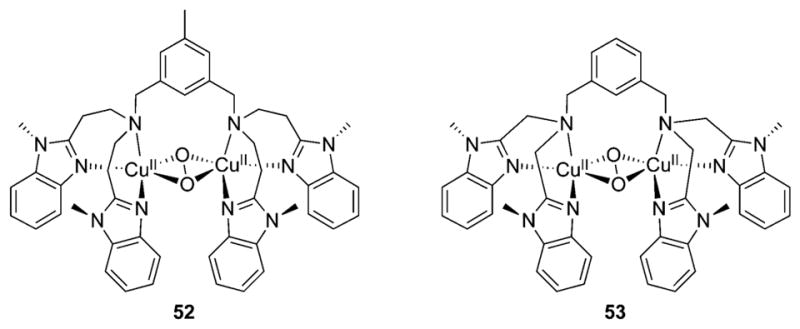
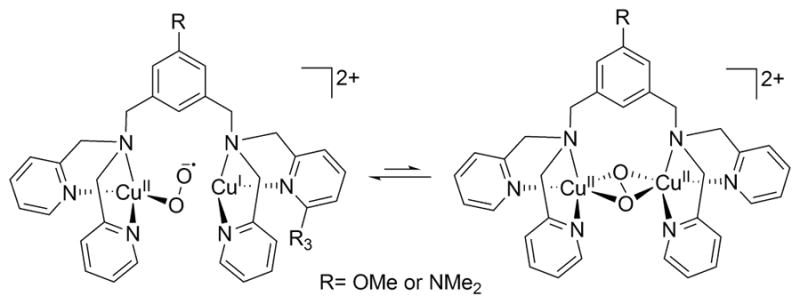
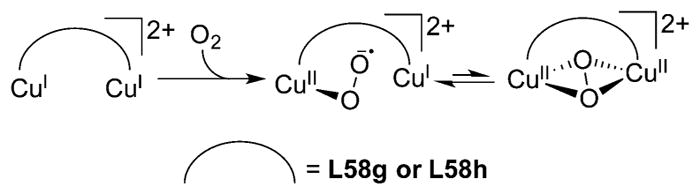
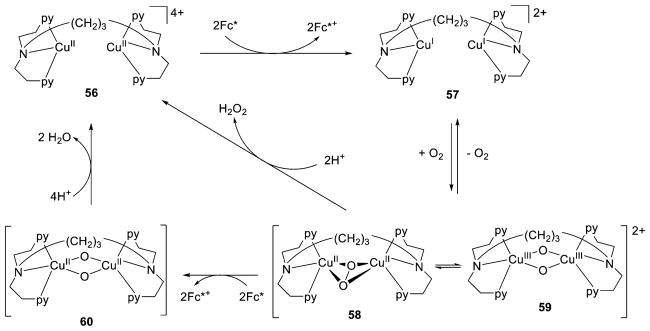
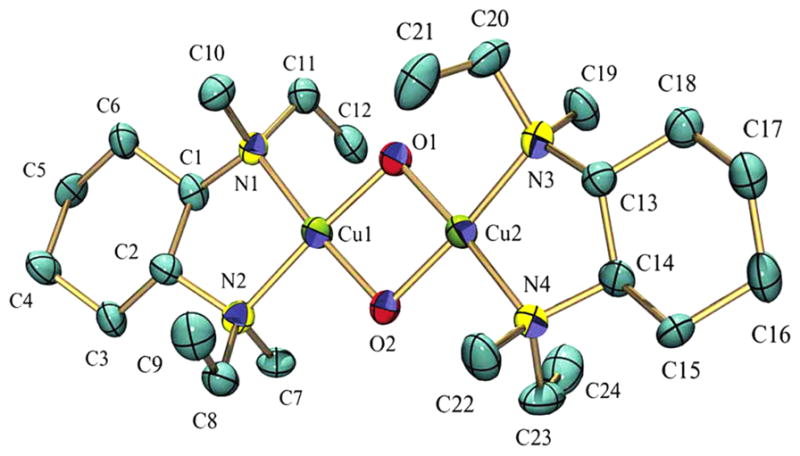
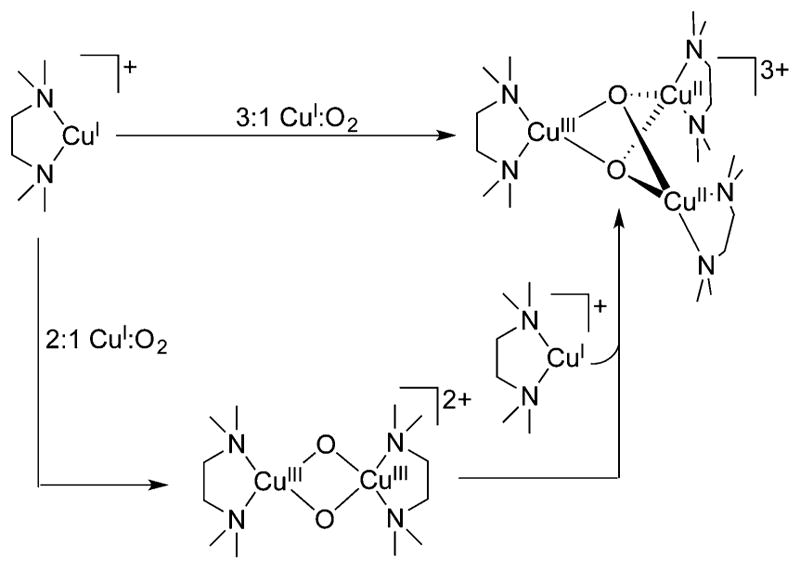
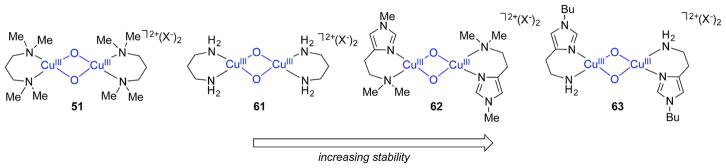
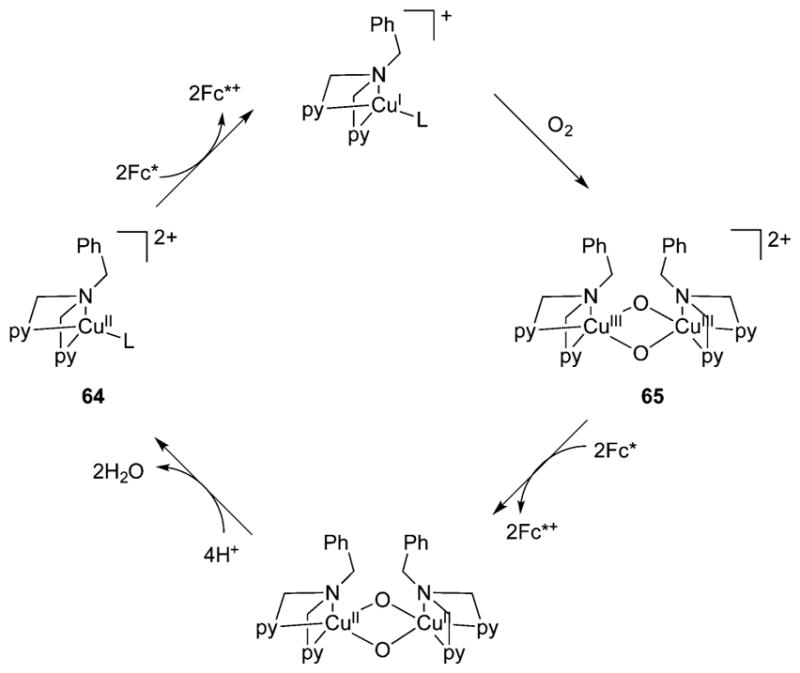
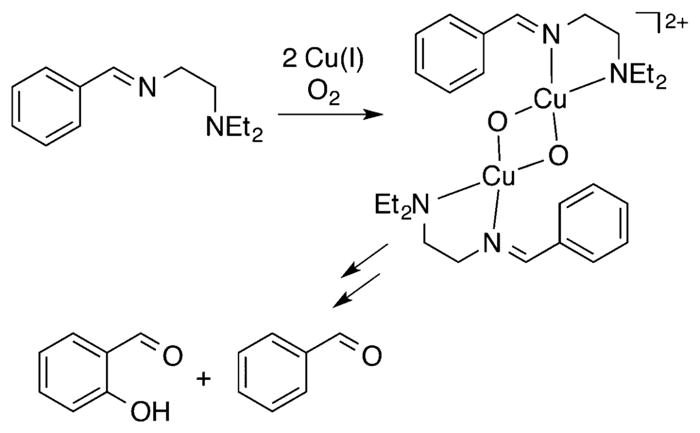
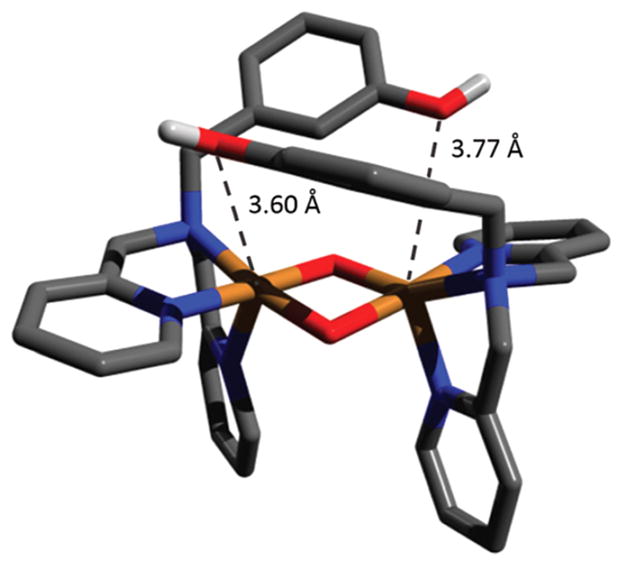
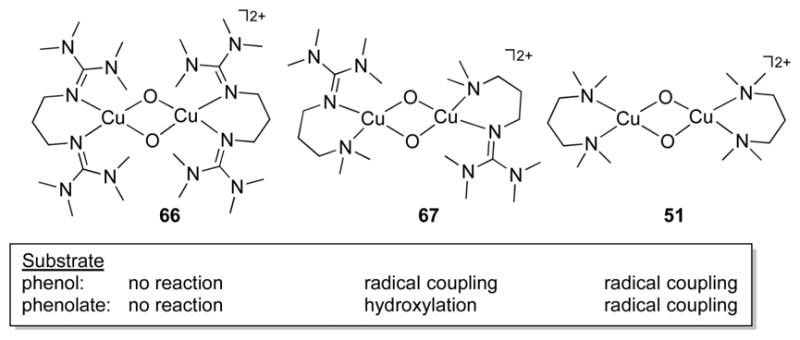

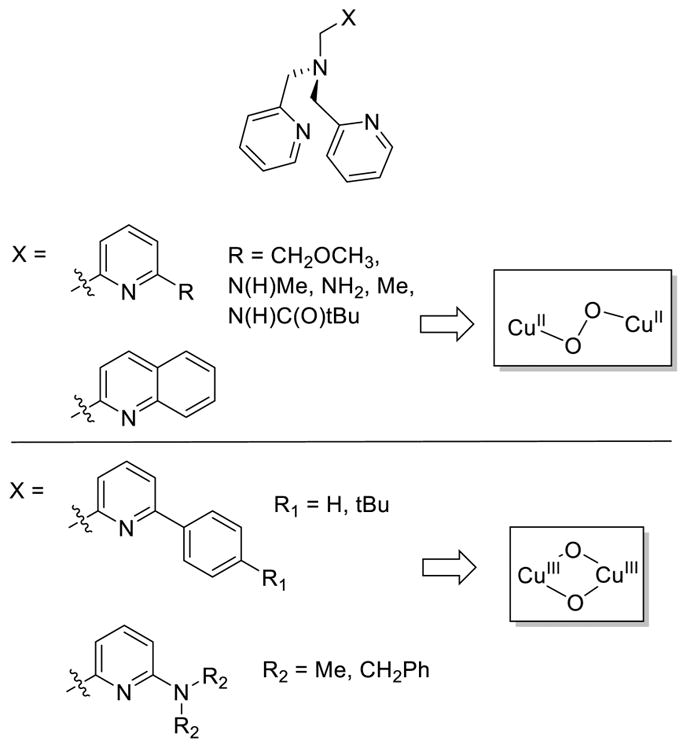
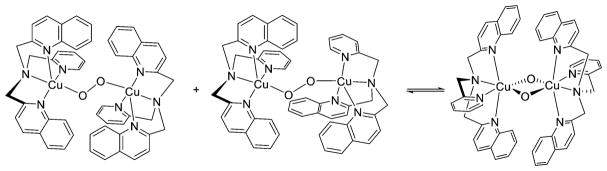
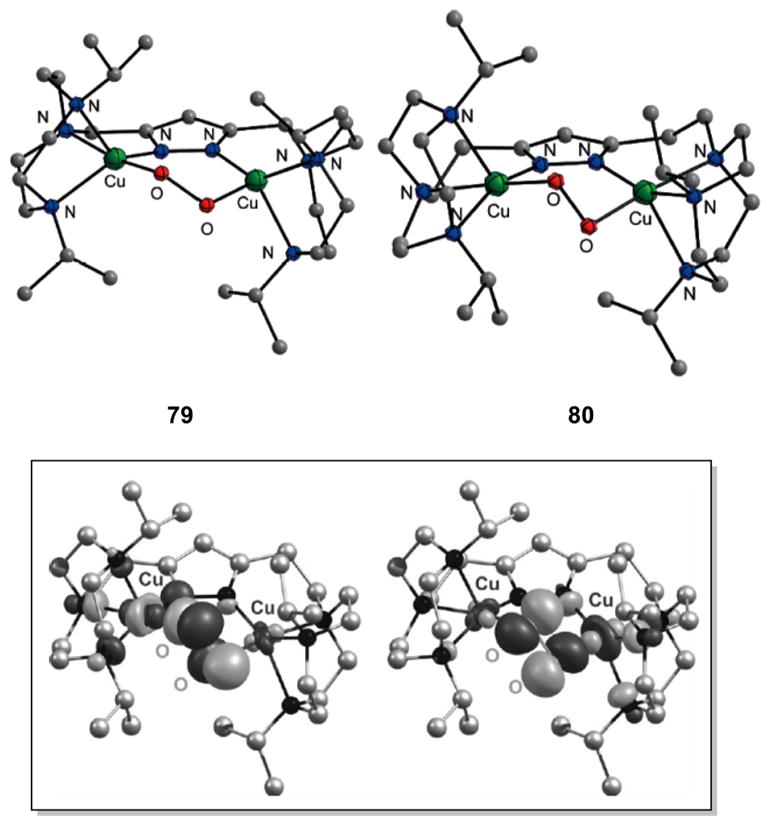
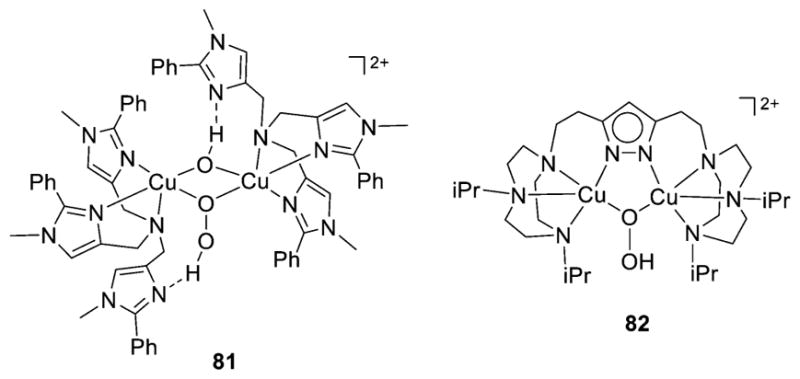
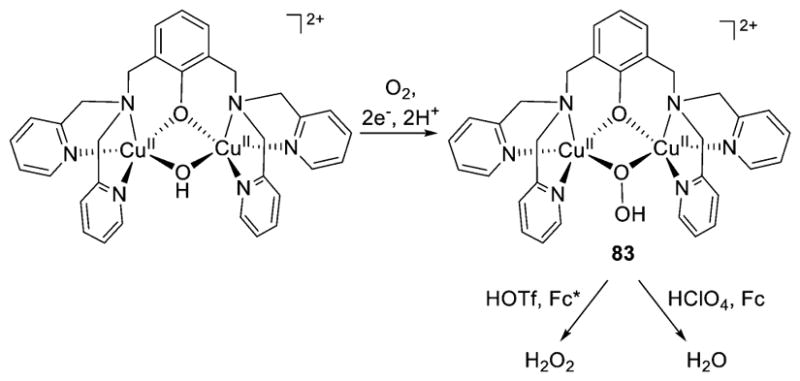
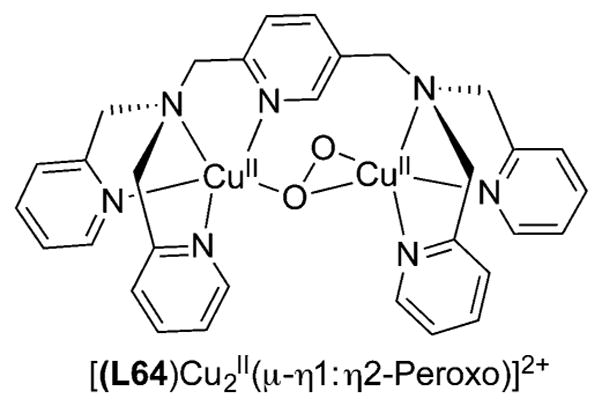
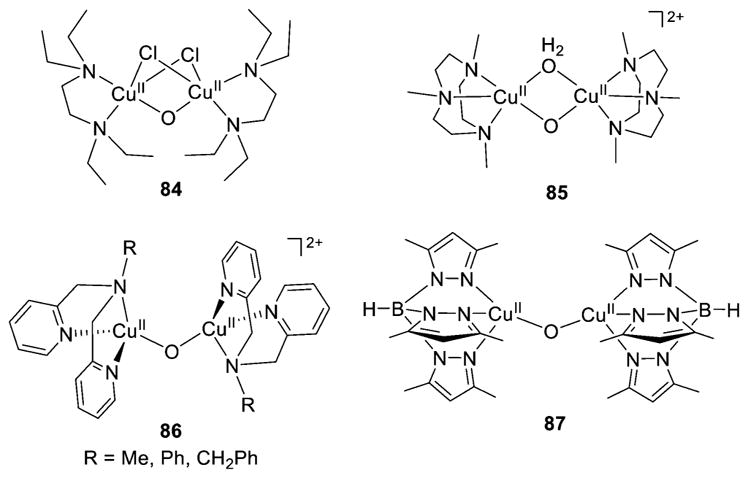
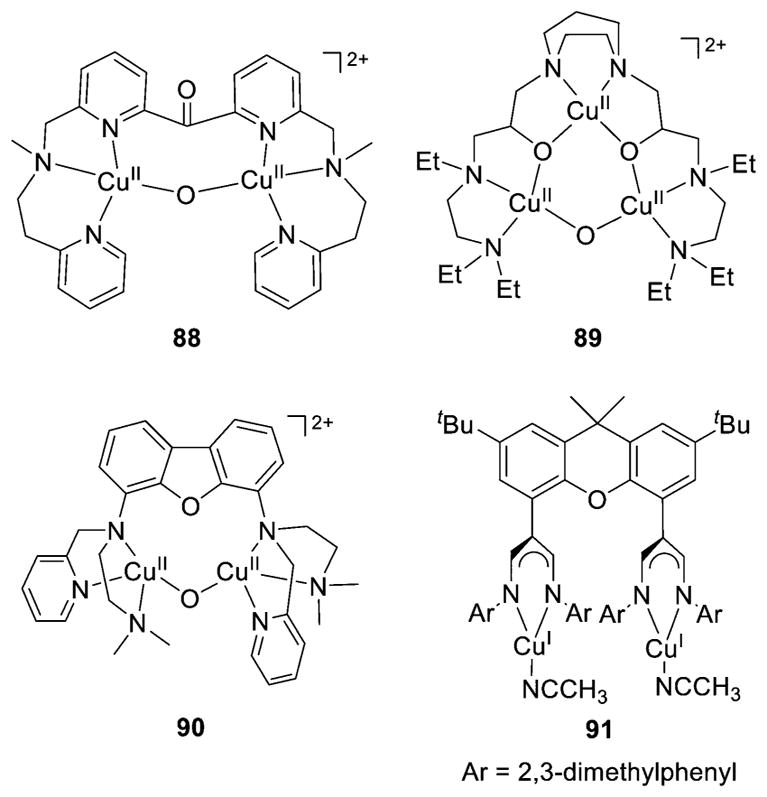

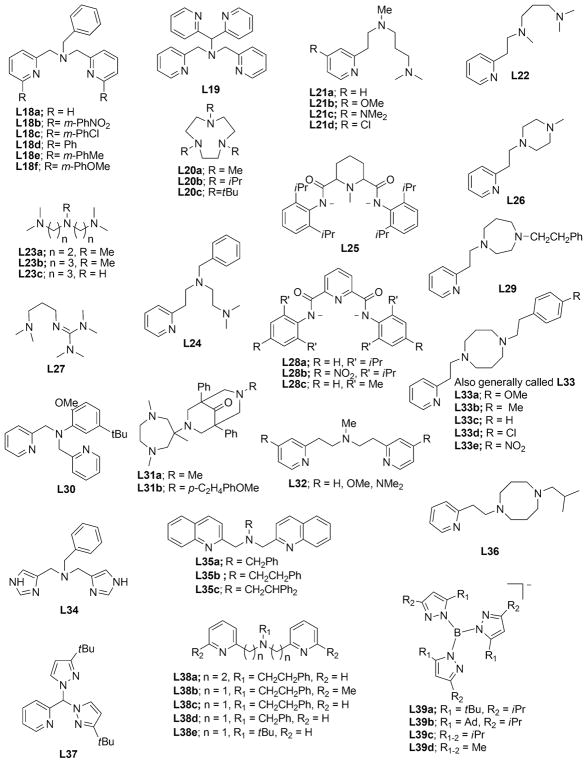
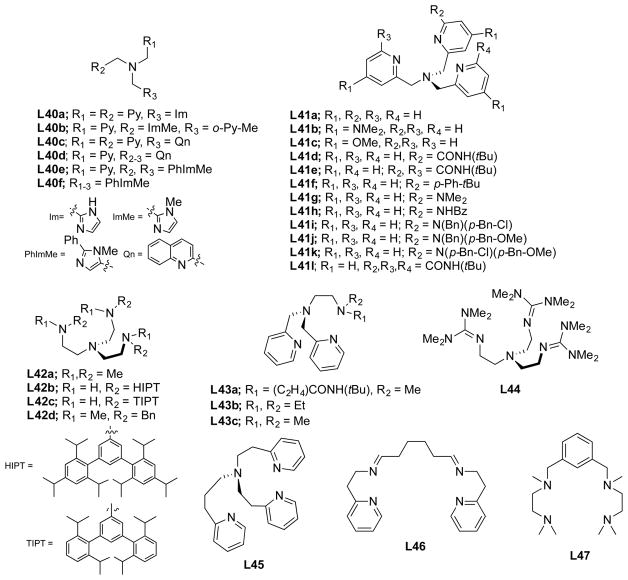
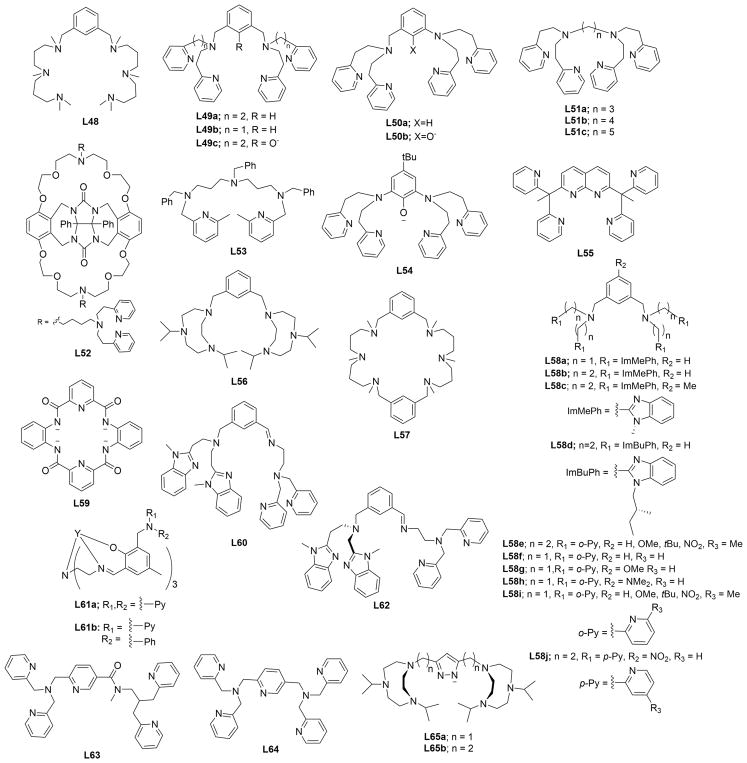
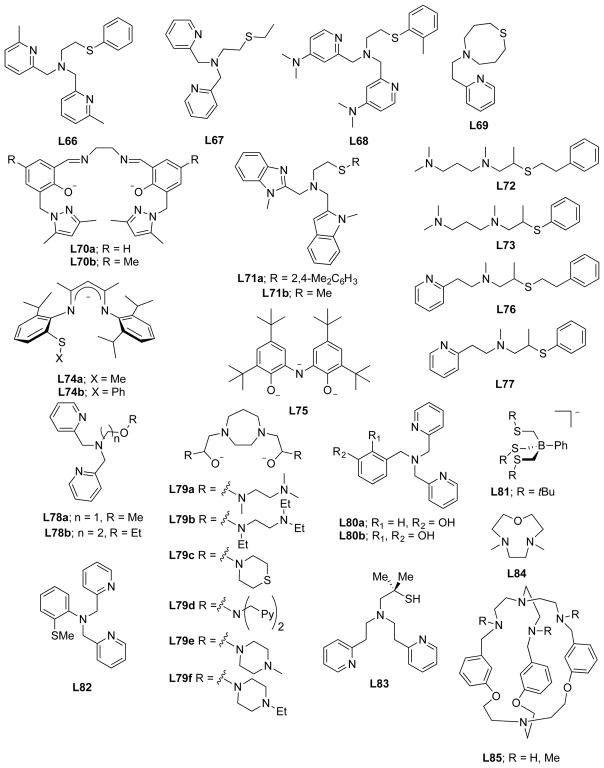
References
-
- Punniyamurthy T, Velusamy S, Iqbal J. Recent Advances in Transition Metal Catalyzed Oxidation of Organic Substrates with Molecular Oxygen. Chem Rev. 2005;105:2329–2364. - PubMed
-
- Wencel-Delord J, Droge T, Liu F, Glorius F. Towards mild metal-catalyzed C-H bond activation. Chem Soc Rev. 2011;40:4740–4761. - PubMed
-
- Yee GM, Tolman WB. Transition Metal Complexes and the Activation of Dioxygen. In: Kroneck PMH, Sosa Torres ME, Sigel A, Sigel H, Sigel RKO, editors. Sustaining Life on Planet Earth: Metalloenzymes Mastering Dioxygen and Other Chewy Gases. Vol. 15. Springer; Cham, Switzerland: 2015. pp. 131–204. Metal Ions in Life Sciences Series.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
