

PARK2-dependent mitophagy induced by acidic postconditioning protects against focal cerebral ischemia and extends the reperfusion window
- PMID: 28103118
- PMCID: PMC5361599
- DOI: 10.1080/15548627.2016.1274596
PARK2-dependent mitophagy induced by acidic postconditioning protects against focal cerebral ischemia and extends the reperfusion window
Abstract
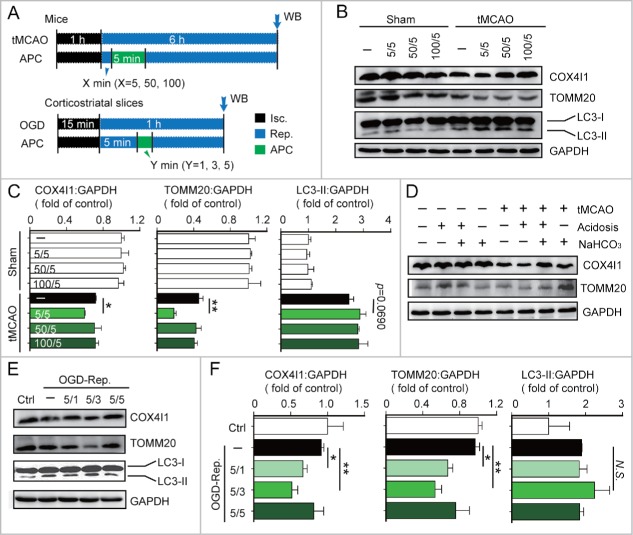
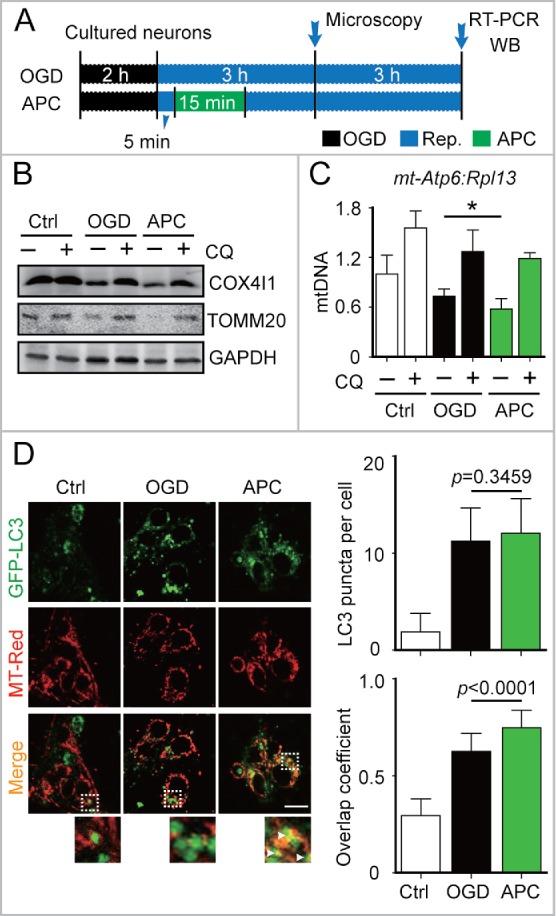
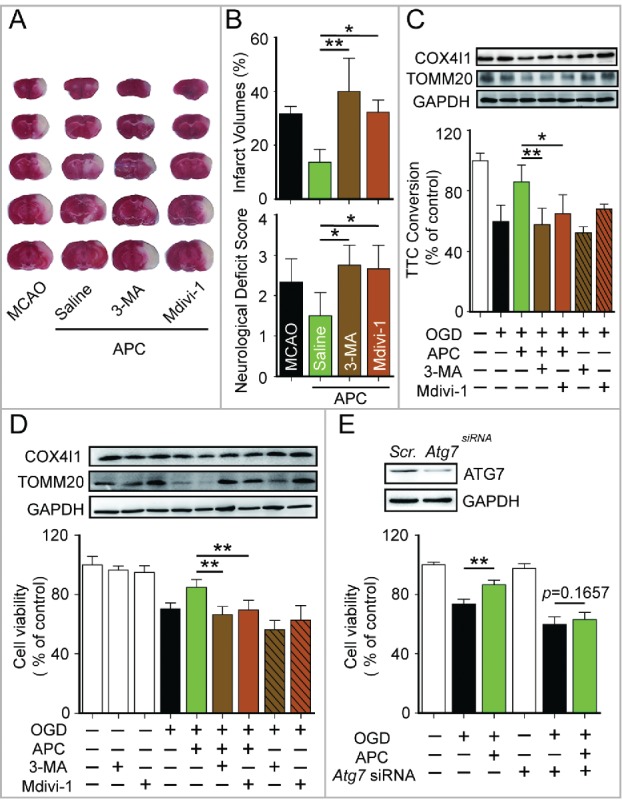
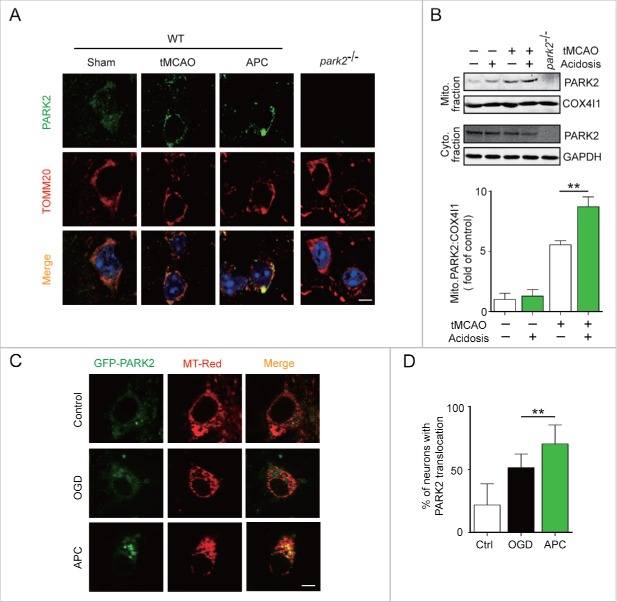
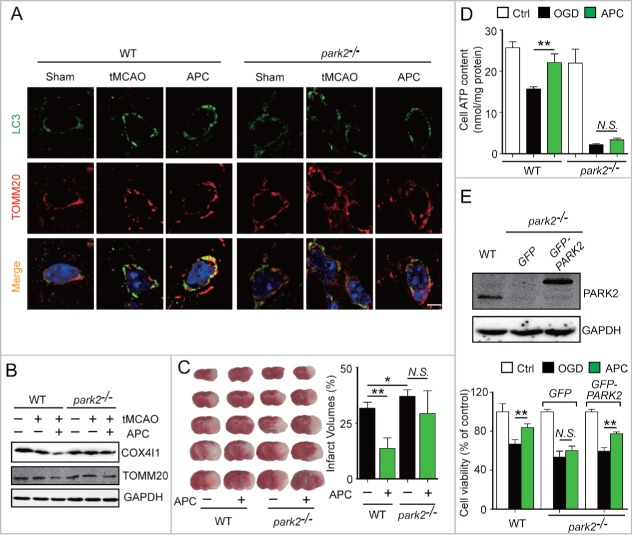
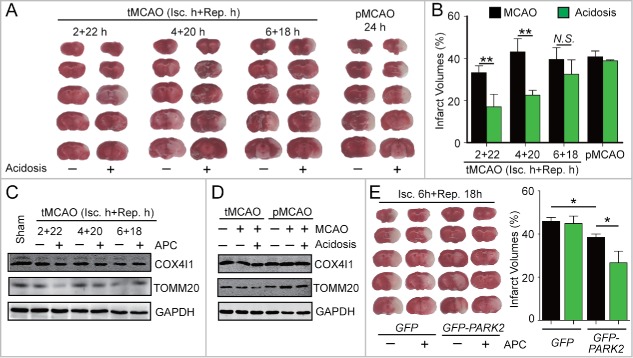
Prompt reperfusion after cerebral ischemia is critical for neuronal survival. Any strategies that extend the limited reperfusion window will be of great importance. Acidic postconditioning (APC) is a mild acidosis treatment that involves inhaling CO2 during reperfusion following ischemia. APC attenuates ischemic brain injury although the underlying mechanisms have not been elucidated. Here we report that APC reinforces ischemia-reperfusion-induced mitophagy in middle cortical artery occlusion (MCAO)-treated mice, and in oxygen-glucose deprivation (OGD)-treated brain slices and neurons. Inhibition of mitophagy compromises neuroprotection conferred by APC. Furthermore, mitophagy and neuroprotection are abolished in Park2 knockout mice, indicating that APC-induced mitophagy is facilitated by the recruitment of PARK2 to mitochondria. Importantly, in MCAO mice, APC treatment extended the effective reperfusion window from 2 to 4 h, and this window was further extended to 6 h by exogenously expressing PARK2. Taken together, we found that PARK2-dependent APC-induced mitophagy renders the brain resistant to ischemic injury. APC treatment could be a favorable strategy to extend the thrombolytic time window for stroke therapy.
Keywords: PARK2; acidic postconditioning; cerebral ischemia; mitophagy; neuroprotection; time window.
Figures
Similar articles
-
Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance.Autophagy. 2013 Sep;9(9):1321-33. doi: 10.4161/auto.25132. Epub 2013 Jun 12. Autophagy. 2013. PMID: 23800795
-
Experimental Models to Study the Neuroprotection of Acidic Postconditioning Against Cerebral Ischemia.J Vis Exp. 2017 Jul 31;(125):55931. doi: 10.3791/55931. J Vis Exp. 2017. PMID: 28784980 Free PMC article.
-
Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy.Autophagy. 2014 Oct 1;10(10):1801-13. doi: 10.4161/auto.32136. Epub 2014 Aug 5. Autophagy. 2014. PMID: 25126734 Free PMC article.
-
Mitochondrial Quality Control in Cerebral Ischemia-Reperfusion Injury.Mol Neurobiol. 2021 Oct;58(10):5253-5271. doi: 10.1007/s12035-021-02494-8. Epub 2021 Jul 18. Mol Neurobiol. 2021. PMID: 34275087 Review.
-
Advances in research of the neuroprotective mechanisms of cerebral ischemic postconditioning.Int J Neurosci. 2015 Mar;125(3):161-9. doi: 10.3109/00207454.2014.917413. Epub 2014 May 23. Int J Neurosci. 2015. PMID: 24754439 Review.
Cited by
-
The Role of Nrf2 in Relieving Cerebral Ischemia-Reperfusion Injury.Curr Neuropharmacol. 2023;21(6):1405-1420. doi: 10.2174/1570159X21666221129100308. Curr Neuropharmacol. 2023. PMID: 36453490 Free PMC article. Review.
-
The Role of Autophagy and Mitophagy in Bone Metabolic Disorders.Int J Biol Sci. 2020 Jul 30;16(14):2675-2691. doi: 10.7150/ijbs.46627. eCollection 2020. Int J Biol Sci. 2020. PMID: 32792864 Free PMC article. Review.
-
Detrimental and Beneficial Effect of Autophagy and a Potential Therapeutic Target after Ischemic Stroke.Evid Based Complement Alternat Med. 2020 Sep 23;2020:8372647. doi: 10.1155/2020/8372647. eCollection 2020. Evid Based Complement Alternat Med. 2020. PMID: 33688357 Free PMC article. Review.
-
Regulated cell death in hypoxic-ischaemic encephalopathy: recent development and mechanistic overview.Cell Death Discov. 2024 Jun 11;10(1):277. doi: 10.1038/s41420-024-02014-2. Cell Death Discov. 2024. PMID: 38862503 Free PMC article. Review.
-
Exercise training alleviates neuronal apoptosis and re-establishes mitochondrial quality control after cerebral ischemia by increasing SIRT3 expression.Cell Biol Toxicol. 2024 Dec 21;41(1):10. doi: 10.1007/s10565-024-09957-3. Cell Biol Toxicol. 2024. PMID: 39707047 Free PMC article.
References
-
- Moskowitz MA. Brain protection: maybe yes, maybe no. Stroke 2010; 41:S85-6; PMID:20876513; http://dx.doi.org/10.1161/STROKEAHA.110.598458 - DOI - PubMed
-
- Del Zoppo GJ, Saver JL, Jauch EC, Adams HP Jr, American Heart Association Stroke C . Expansion of the time window for treatment of acute ischemic stroke with intravenous tissue plasminogen activator: a science advisory from the American Heart Association/American Stroke Association. Stroke 2009; 40:2945-8; PMID:19478221; http://dx.doi.org/10.1161/STROKEAHA.109.192535 - DOI - PMC - PubMed
-
- Zhao H, Sapolsky RM, Steinberg GK. Interrupting reperfusion as a stroke therapy: ischemic postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metab 2006; 26:1114-21; PMID:16736038; http://dx.doi.org/10.1038/sj.jcbfm.9600233 - DOI - PubMed
-
- Leconte C, Tixier E, Freret T, Toutain J, Saulnier R, Boulouard M, Roussel S, Schumann-Bard P, Bernaudin M. Delayed hypoxic postconditioning protects against cerebral ischemia in the mouse. Stroke 2009; 40:3349-55; PMID:19628803; http://dx.doi.org/10.1161/STROKEAHA.109.557314 - DOI - PubMed
-
- Fan YY, Zhang XN, He P, Shen Z, Shen Y, Wang XF, Hu WW, Chen Z. Transient lack of glucose but not O2 is involved in ischemic postconditioning-induced neuroprotection. CNS Neuroscience Therapeutics 2013; 19:30-7; http://dx.doi.org/10.1111/cns.12033 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources