

Motor Learning Requires Purkinje Cell Synaptic Potentiation through Activation of AMPA-Receptor Subunit GluA3
- PMID: 28103481
- PMCID: PMC5263704
- DOI: 10.1016/j.neuron.2016.11.046
Motor Learning Requires Purkinje Cell Synaptic Potentiation through Activation of AMPA-Receptor Subunit GluA3
Abstract
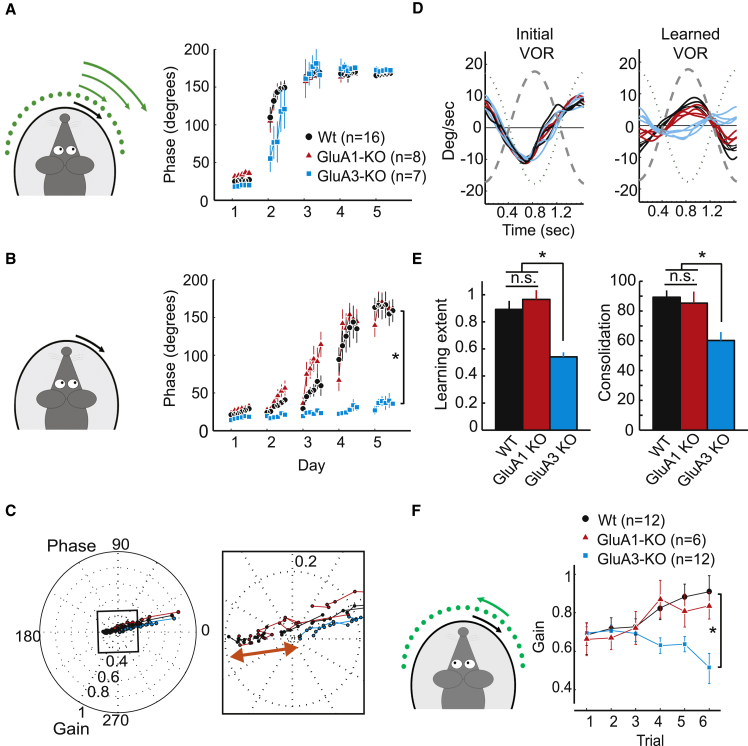
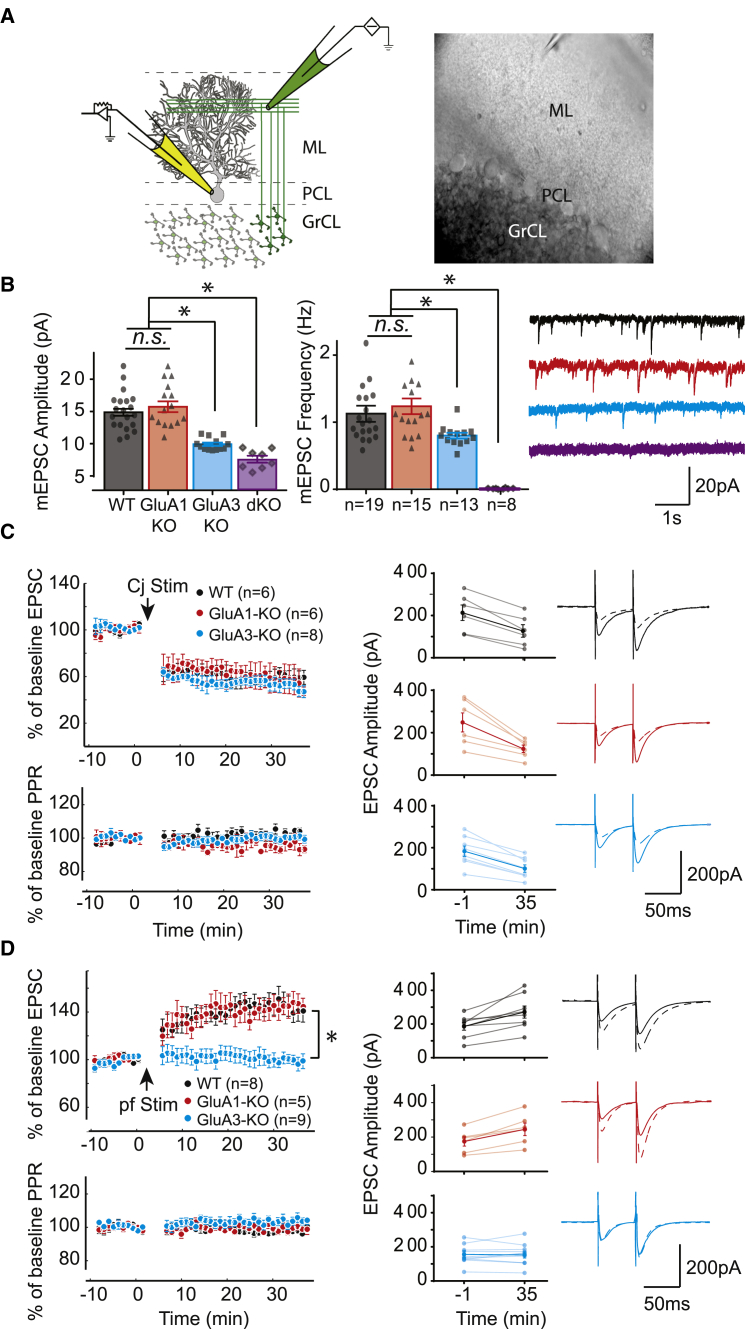
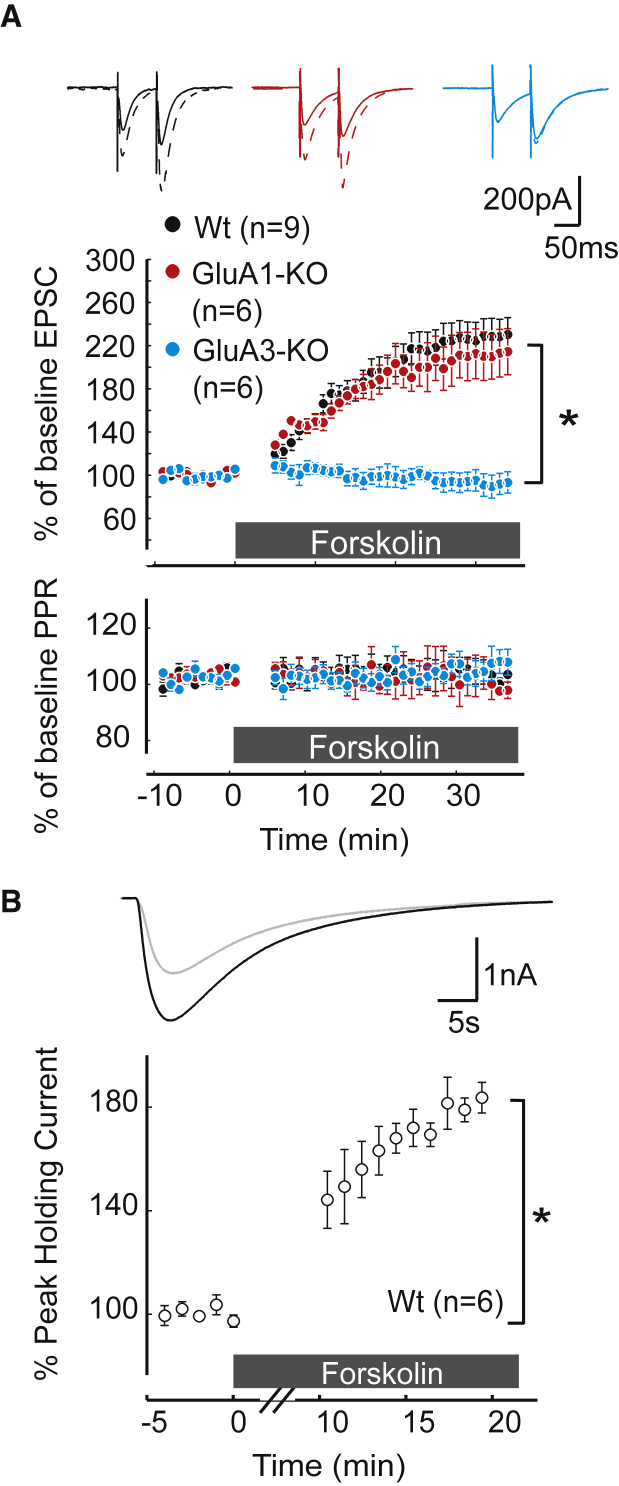
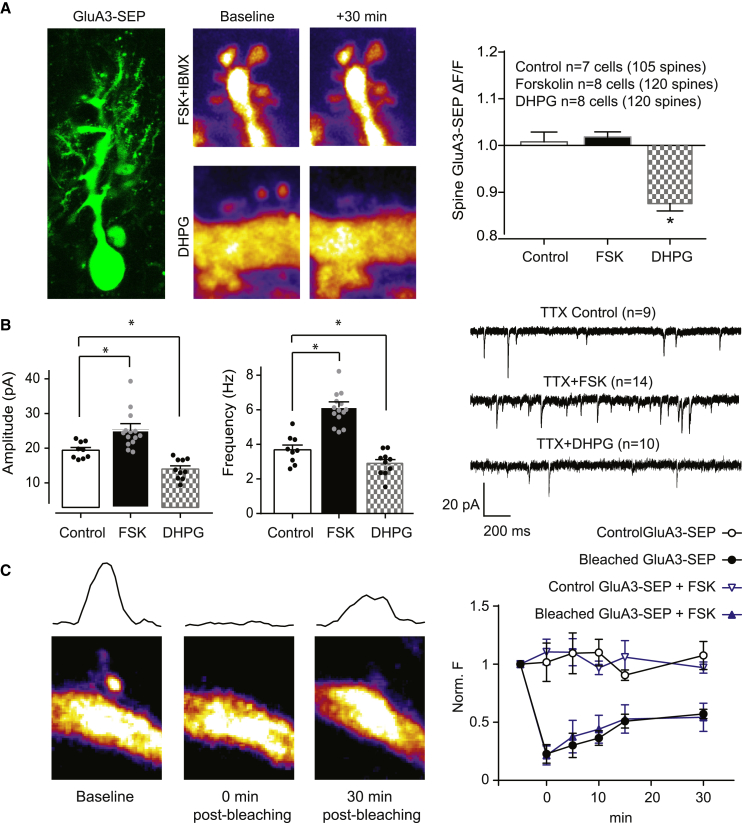
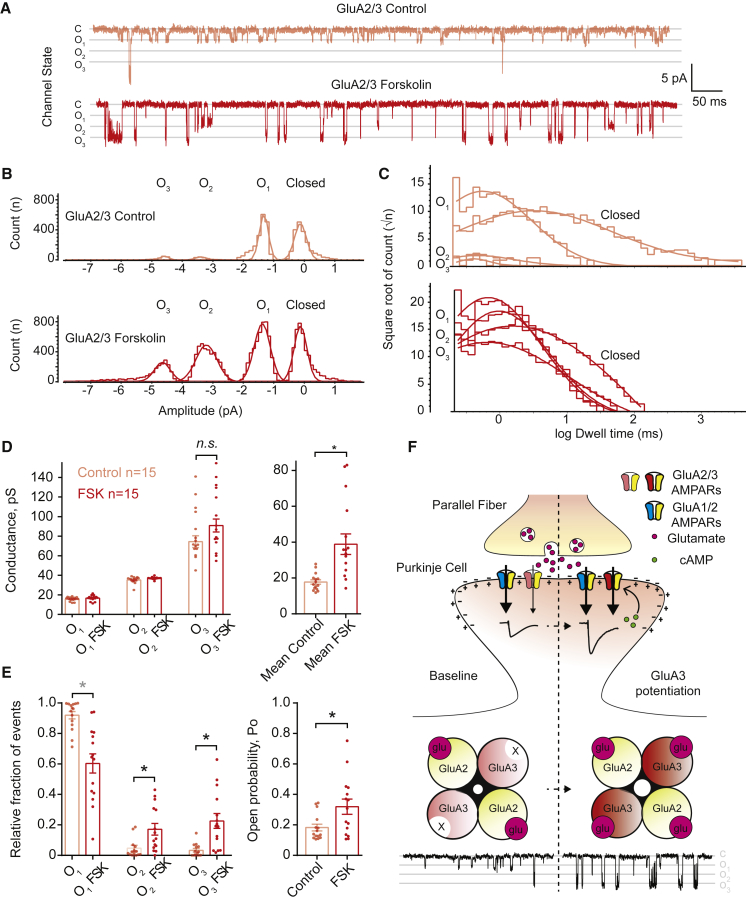
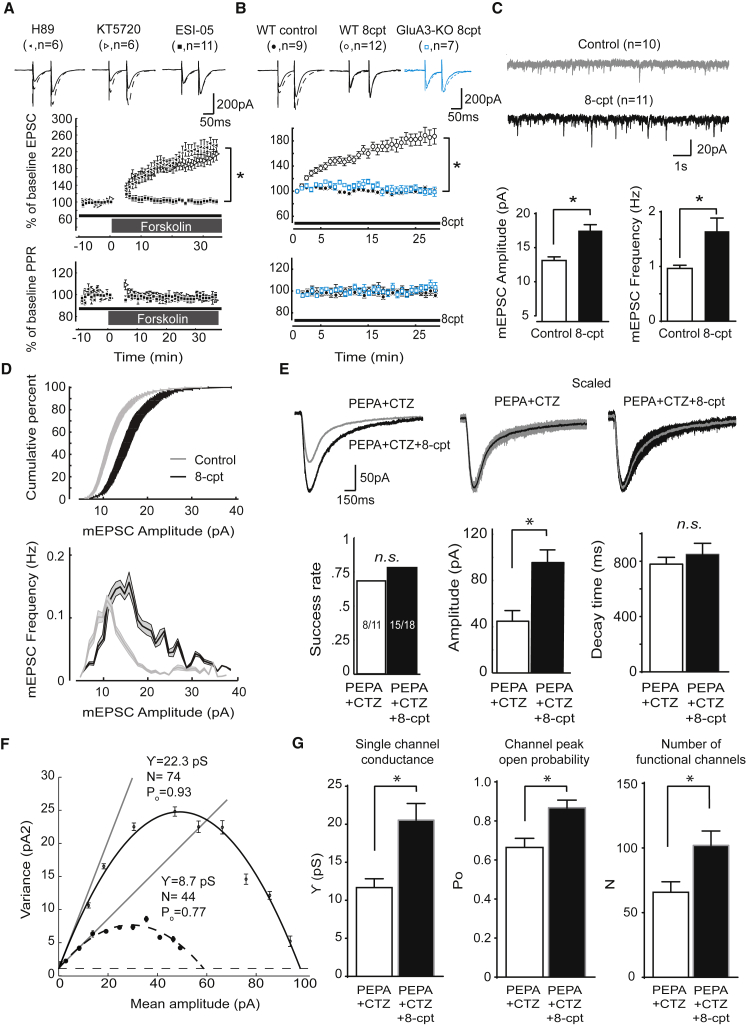
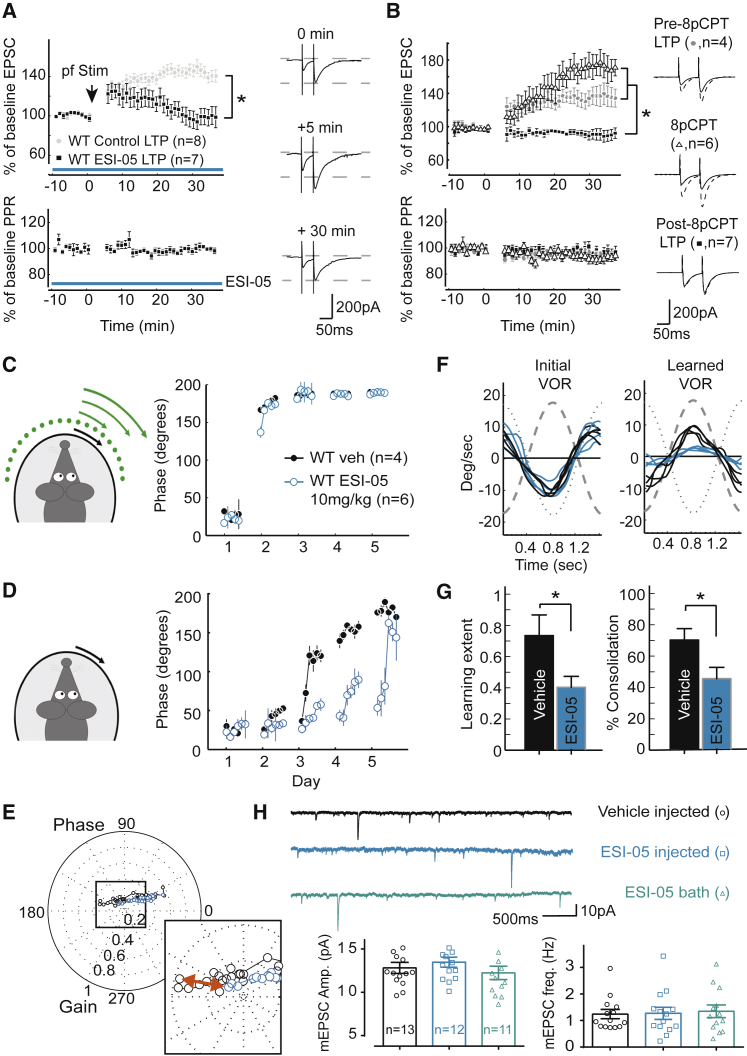
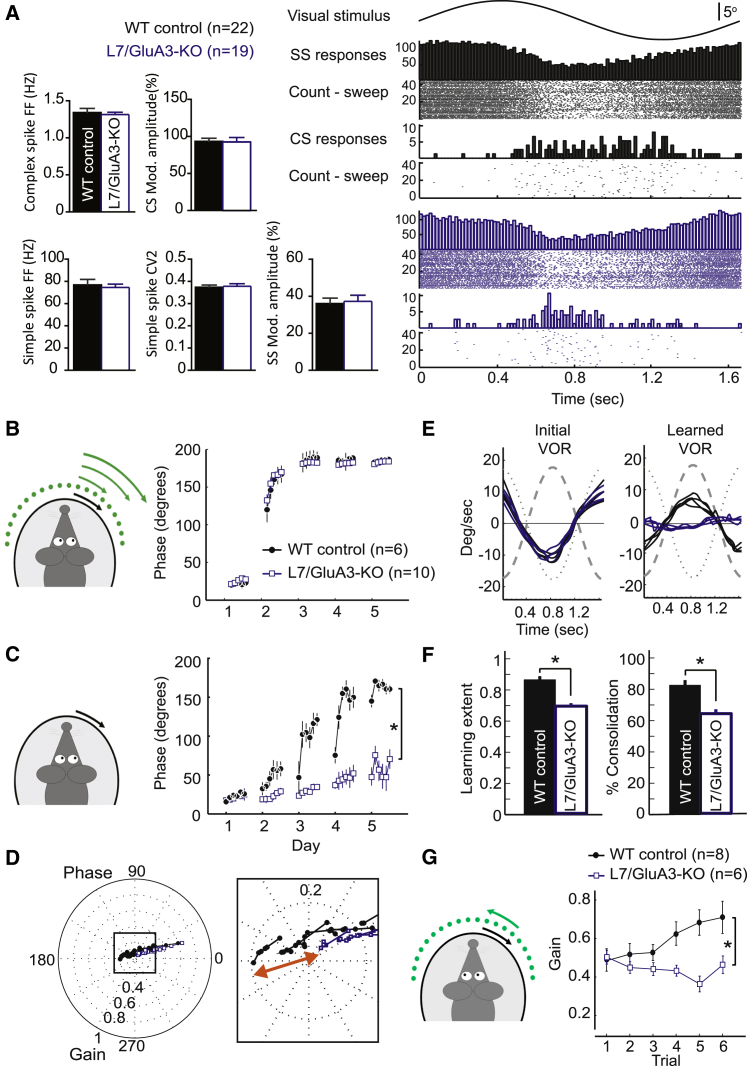
Accumulating evidence indicates that cerebellar long-term potentiation (LTP) is necessary for procedural learning. However, little is known about its underlying molecular mechanisms. Whereas AMPA receptor (AMPAR) subunit rules for synaptic plasticity have been extensively studied in relation to declarative learning, it is unclear whether these rules apply to cerebellum-dependent motor learning. Here we show that LTP at the parallel-fiber-to-Purkinje-cell synapse and adaptation of the vestibulo-ocular reflex depend not on GluA1- but on GluA3-containing AMPARs. In contrast to the classic form of LTP implicated in declarative memory formation, this form of LTP does not require GluA1-AMPAR trafficking but rather requires changes in open-channel probability of GluA3-AMPARs mediated by cAMP signaling and activation of the protein directly activated by cAMP (Epac). We conclude that vestibulo-cerebellar motor learning is the first form of memory acquisition shown to depend on GluA3-dependent synaptic potentiation by increasing single-channel conductance.
Keywords: AMPA receptor; Epac; GluA3; LTP; Purkinje cell; cerebellum; learning; synapse.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Anzai M., Kitazawa H., Nagao S. Effects of reversible pharmacological shutdown of cerebellar flocculus on the memory of long-term horizontal vestibulo-ocular reflex adaptation in monkeys. Neurosci. Res. 2010;68:191–198. - PubMed
-
- Barski J.J., Dethleffsen K., Meyer M. Cre recombinase expression in cerebellar Purkinje cells. Genesis. 2000;28:93–98. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
