

Proteomics informed by transcriptomics for characterising active transposable elements and genome annotation in Aedes aegypti
- PMID: 28103802
- PMCID: PMC5248466
- DOI: 10.1186/s12864-016-3432-5
Proteomics informed by transcriptomics for characterising active transposable elements and genome annotation in Aedes aegypti
Abstract
Background: Aedes aegypti is a vector for the (re-)emerging human pathogens dengue, chikungunya, yellow fever and Zika viruses. Almost half of the Ae. aegypti genome is comprised of transposable elements (TEs). Transposons have been linked to diverse cellular processes, including the establishment of viral persistence in insects, an essential step in the transmission of vector-borne viruses. However, up until now it has not been possible to study the overall proteome derived from an organism's mobile genetic elements, partly due to the highly divergent nature of TEs. Furthermore, as for many non-model organisms, incomplete genome annotation has hampered proteomic studies on Ae. aegypti.
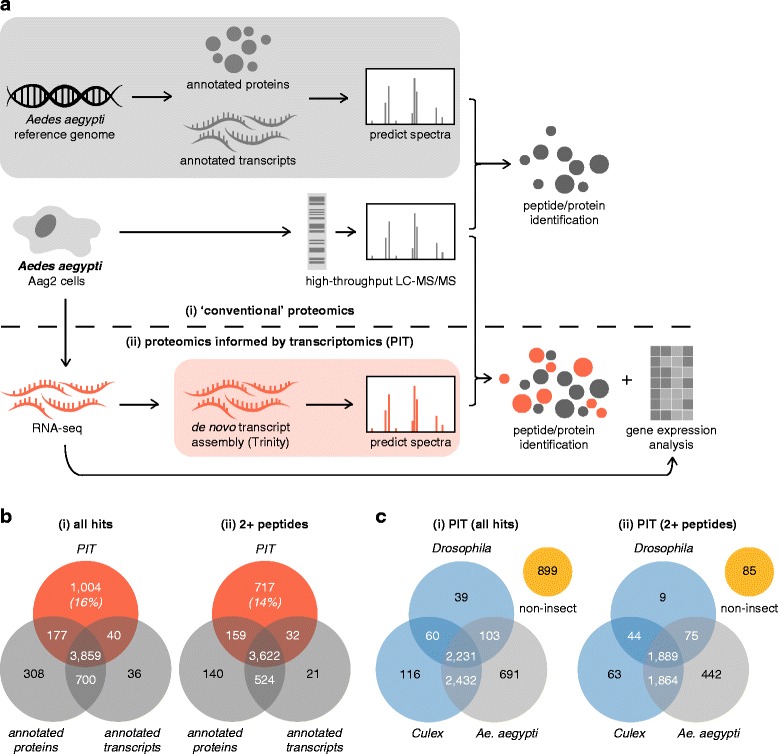
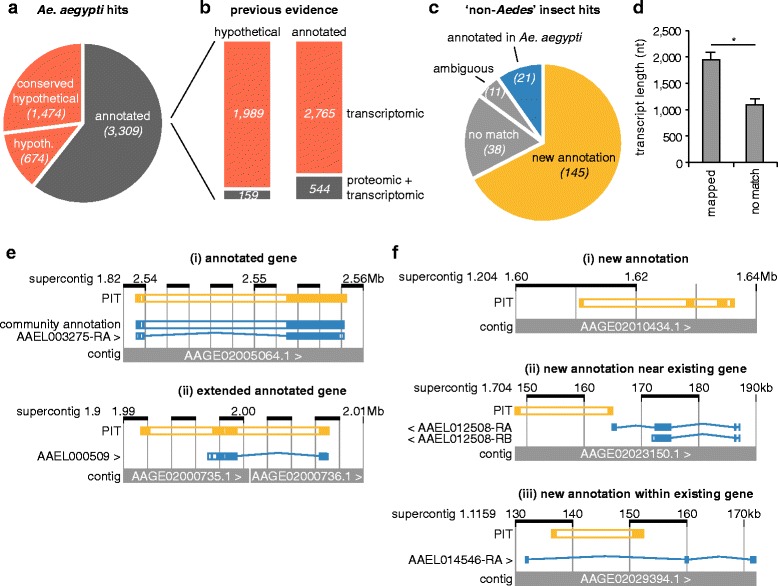
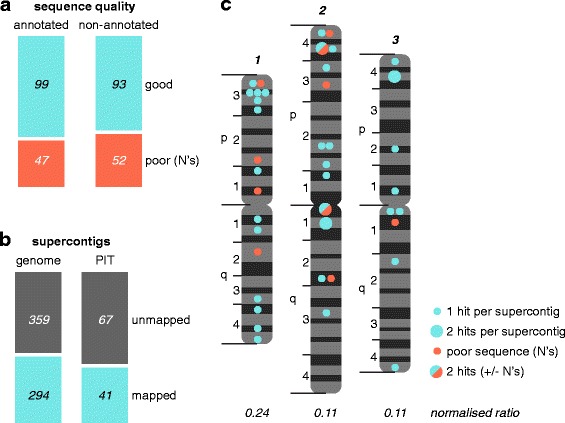
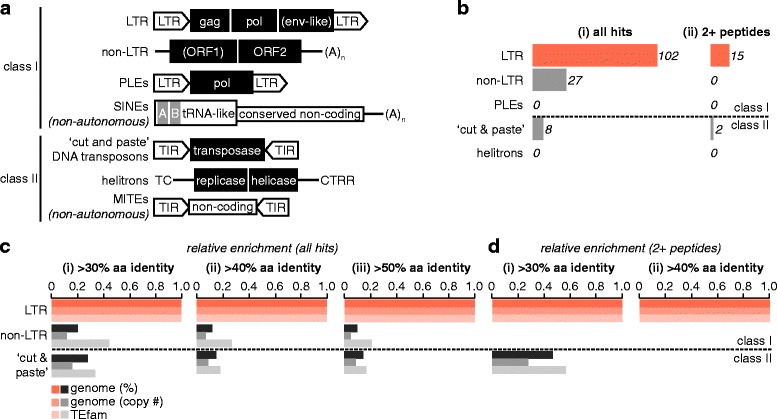
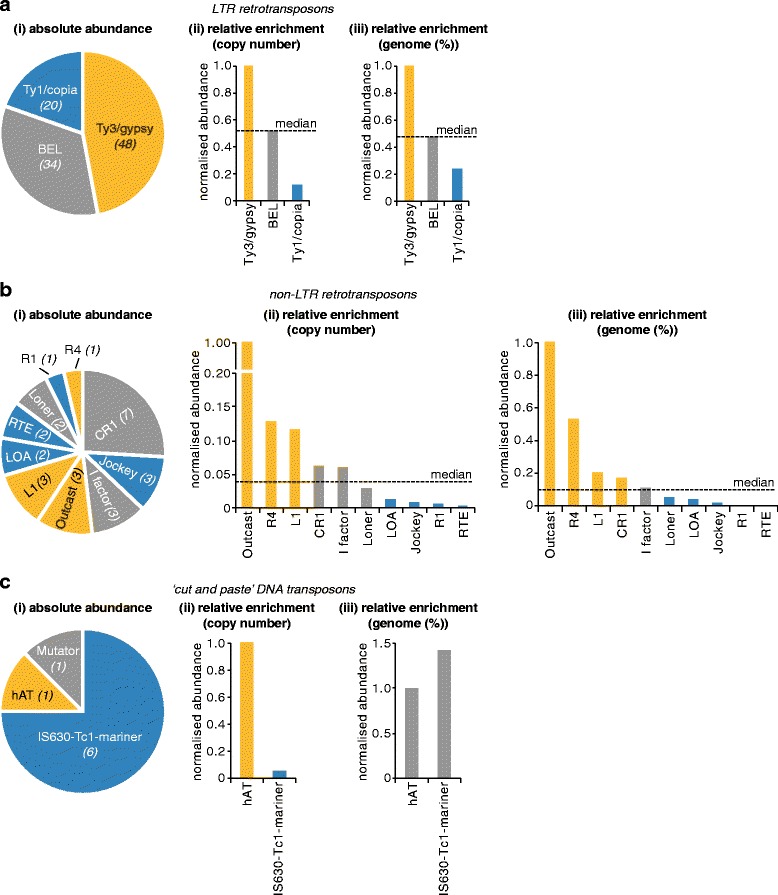
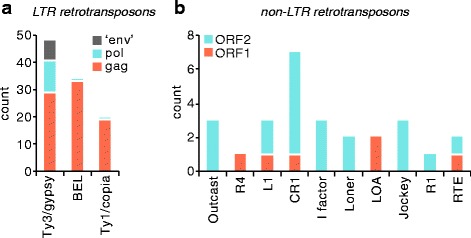
Results: We analysed the Ae. aegypti proteome using our new proteomics informed by transcriptomics (PIT) technique, which bypasses the need for genome annotation by identifying proteins through matched transcriptomic (rather than genomic) data. Our data vastly increase the number of experimentally confirmed Ae. aegypti proteins. The PIT analysis also identified hotspots of incomplete genome annotation, and showed that poor sequence and assembly quality do not explain all annotation gaps. Finally, in a proof-of-principle study, we developed criteria for the characterisation of proteomically active TEs. Protein expression did not correlate with a TE's genomic abundance at different levels of classification. Most notably, long terminal repeat (LTR) retrotransposons were markedly enriched compared to other elements. PIT was superior to 'conventional' proteomic approaches in both our transposon and genome annotation analyses.
Conclusions: We present the first proteomic characterisation of an organism's repertoire of mobile genetic elements, which will open new avenues of research into the function of transposon proteins in health and disease. Furthermore, our study provides a proof-of-concept that PIT can be used to evaluate a genome's annotation to guide annotation efforts which has the potential to improve the efficiency of annotation projects in non-model organisms. PIT therefore represents a valuable new tool to study the biology of the important vector species Ae. aegypti, including its role in transmitting emerging viruses of global public health concern.
Keywords: Aedes aegypti; Genome annotation; Non-model organism; PIT; Proteomics informed by transcriptomics; Transposon.
Figures

Comment in
-
Proteomics technique opens new frontiers in mobilome research.Mob Genet Elements. 2017 Aug 1;7(4):1-9. doi: 10.1080/2159256X.2017.1362494. eCollection 2017. Mob Genet Elements. 2017. PMID: 28932623 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- BB/K016075/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/M02542X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- R01 AI073450/AI/NIAID NIH HHS/United States
- G0801973/MRC_/Medical Research Council/United Kingdom
- BB/M020118/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
