

Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy
- PMID: 28104840
- PMCID: PMC5592794
- DOI: 10.1126/science.aaf8399
Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy
Abstract
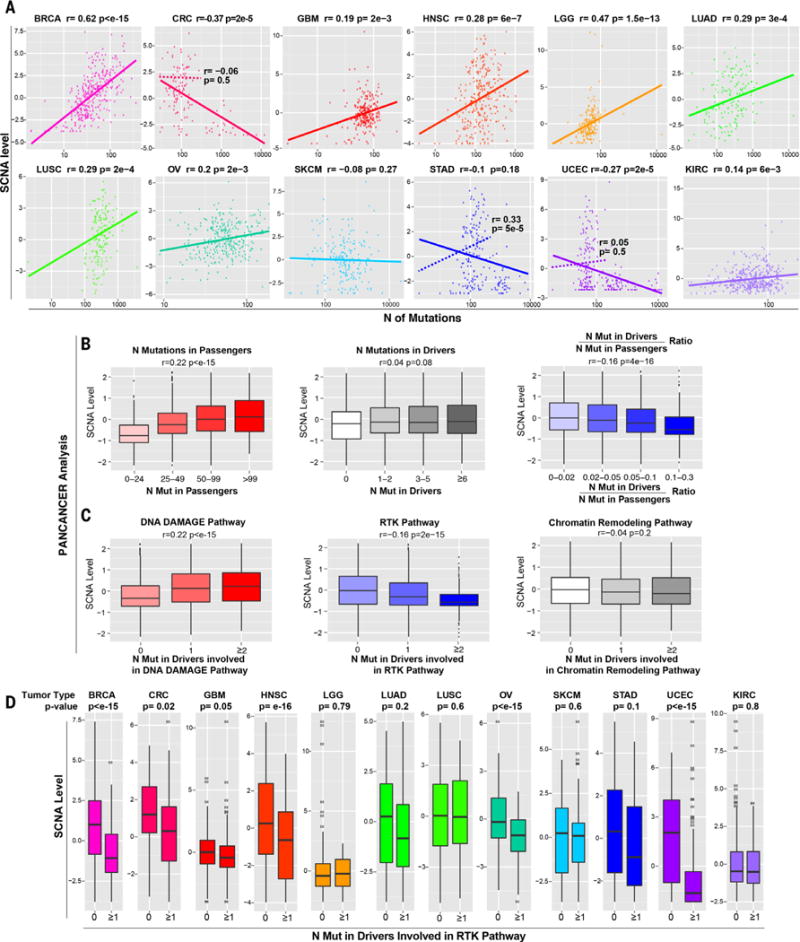
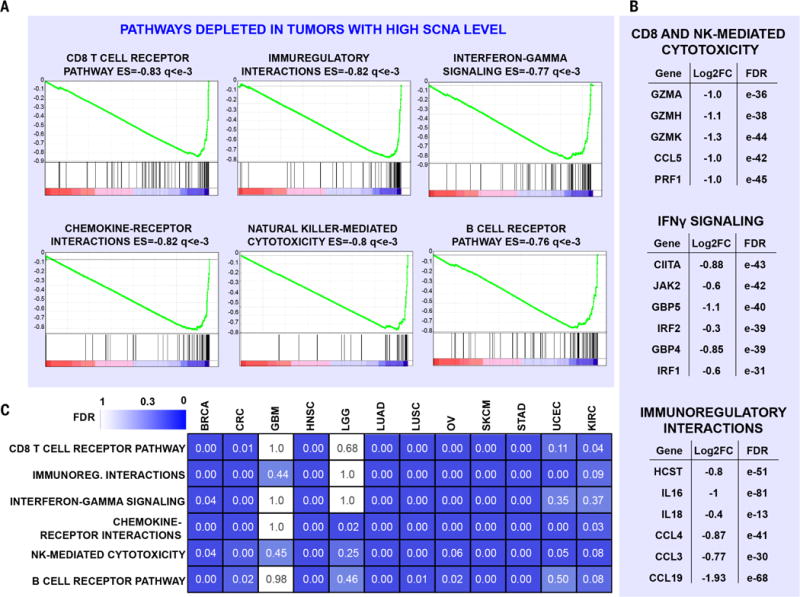
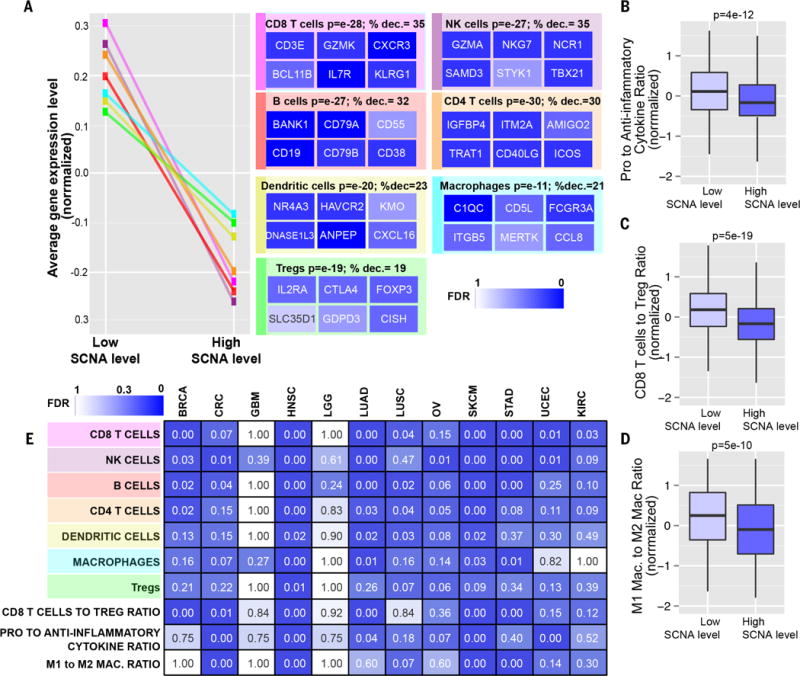
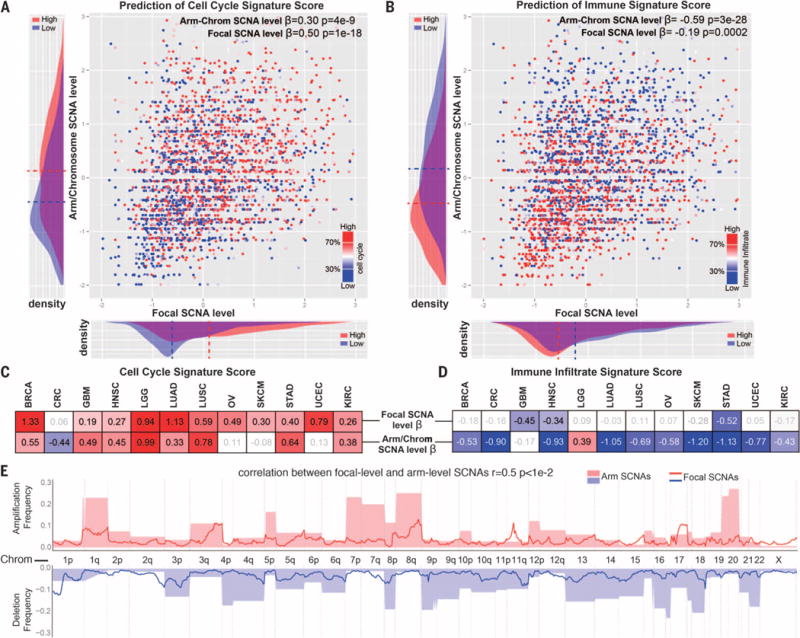
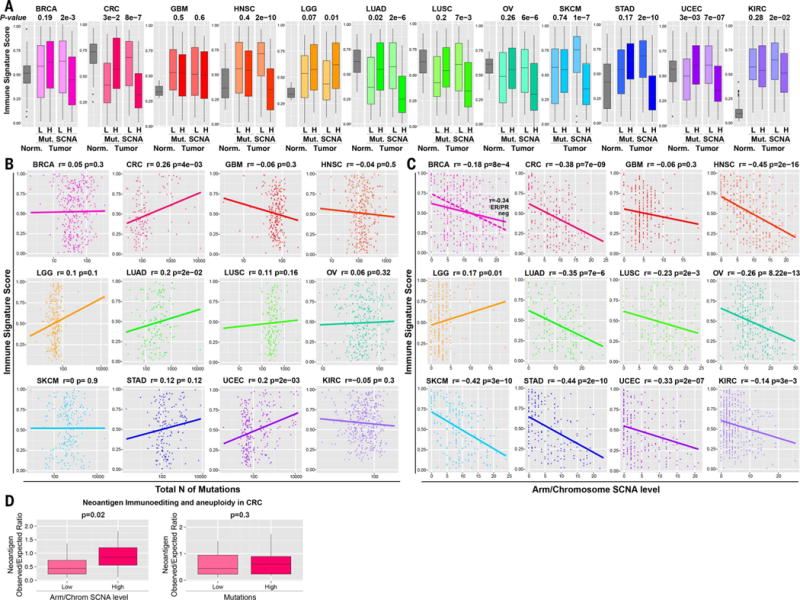
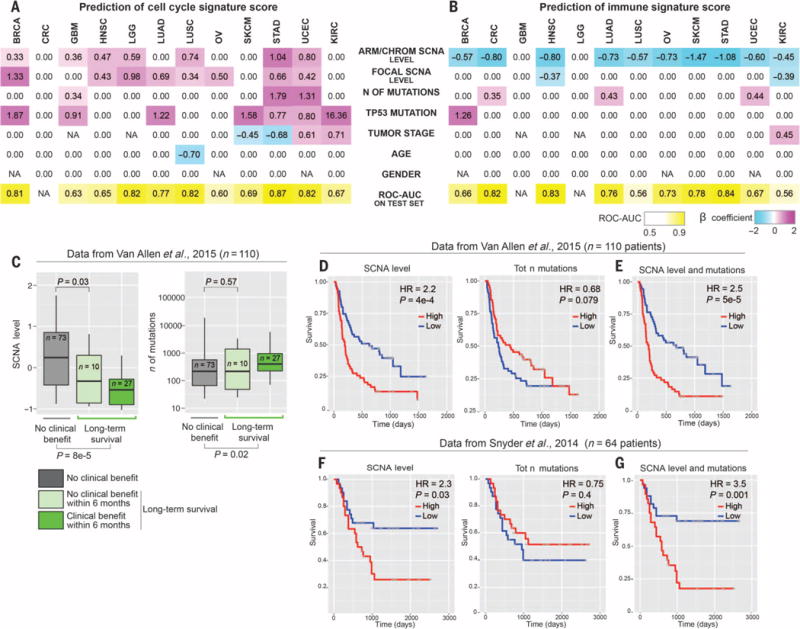
Immunotherapies based on immune checkpoint blockade are highly effective in a subset of patients. An ongoing challenge is the identification of biomarkers that predict which patients will benefit from these therapies. Aneuploidy, also known as somatic copy number alterations (SCNAs), is widespread in cancer and is posited to drive tumorigenesis. Analyzing 12 human cancer types, we find that, for most, highly aneuploid tumors show reduced expression of markers of cytotoxic infiltrating immune cells, especially CD8+ T cells, and increased expression of cell proliferation markers. Different types of SCNAs predict the proliferation and immune signatures, implying distinct underlying mechanisms. Using published data from two clinical trials of immune checkpoint blockade therapy for metastatic melanoma, we found that tumor aneuploidy inversely correlates with patient survival. Together with other tumor characteristics such as tumor mutational load, aneuploidy may thus help identify patients most likely to respond to immunotherapy.
Copyright © 2017, American Association for the Advancement of Science.
Figures
Comment in
-
Chromosomal chaos silences immune surveillance.Science. 2017 Jan 20;355(6322):249-250. doi: 10.1126/science.aam5331. Science. 2017. PMID: 28104855 No abstract available.
-
Biomarkers: Aneuploidy and immune evasion - a biomarker of response.Nat Rev Clin Oncol. 2017 Feb 20;14(3):140. doi: 10.1038/nrclinonc.2017.23. Nat Rev Clin Oncol. 2017. PMID: 28218256 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
