

Protein structure determination using metagenome sequence data
- PMID: 28104891
- PMCID: PMC5493203
- DOI: 10.1126/science.aah4043
Protein structure determination using metagenome sequence data
Abstract
Despite decades of work by structural biologists, there are still ~5200 protein families with unknown structure outside the range of comparative modeling. We show that Rosetta structure prediction guided by residue-residue contacts inferred from evolutionary information can accurately model proteins that belong to large families and that metagenome sequence data more than triple the number of protein families with sufficient sequences for accurate modeling. We then integrate metagenome data, contact-based structure matching, and Rosetta structure calculations to generate models for 614 protein families with currently unknown structures; 206 are membrane proteins and 137 have folds not represented in the Protein Data Bank. This approach provides the representative models for large protein families originally envisioned as the goal of the Protein Structure Initiative at a fraction of the cost.
Copyright © 2017, American Association for the Advancement of Science.
Figures
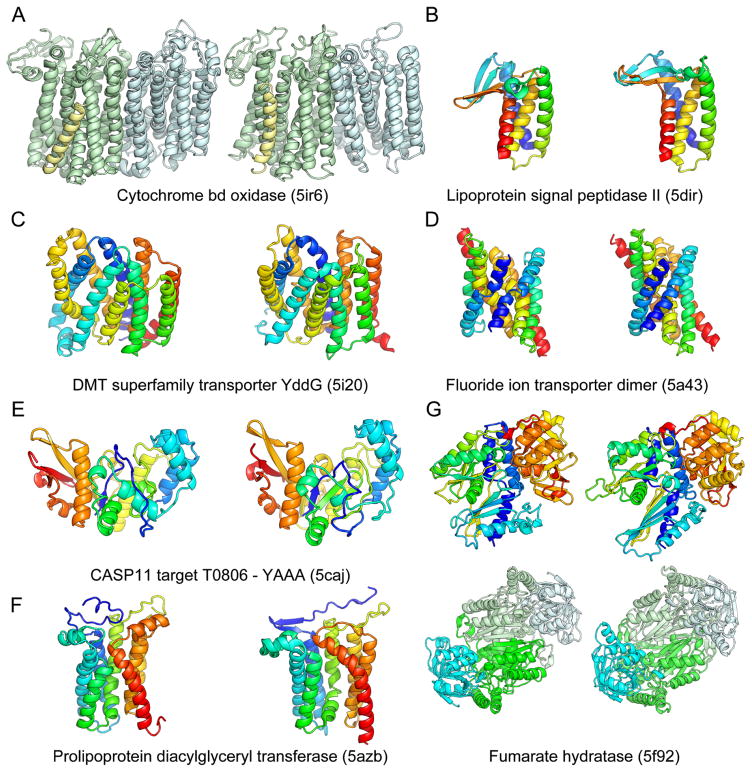
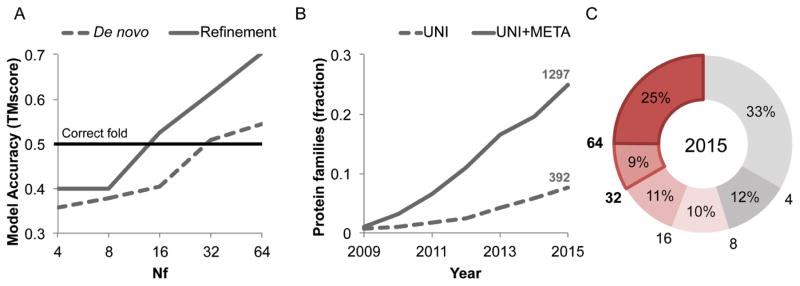
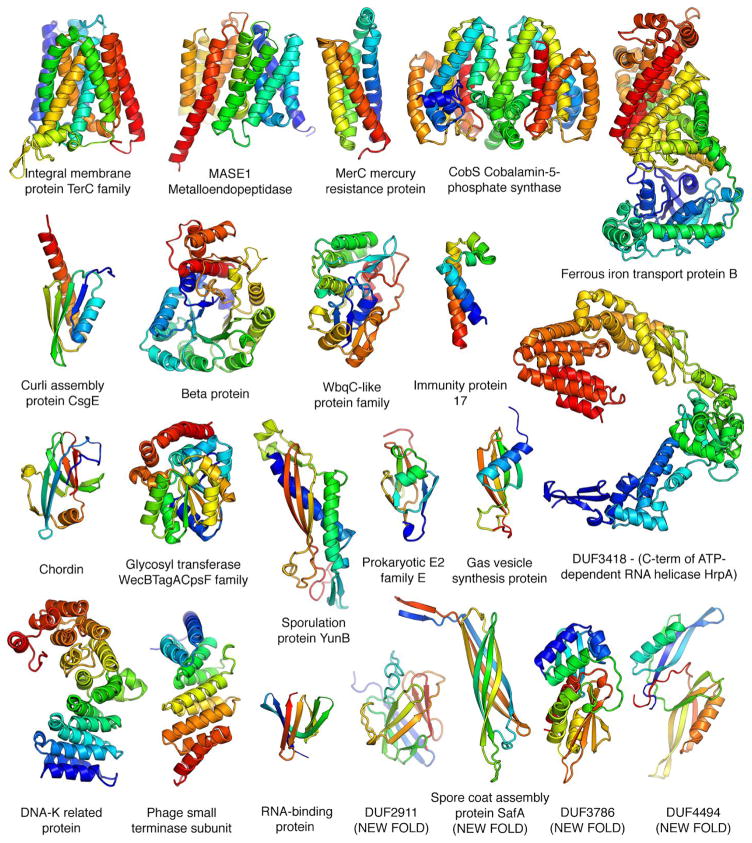
Comment in
-
Big-data approaches to protein structure prediction.Science. 2017 Jan 20;355(6322):248-249. doi: 10.1126/science.aal4512. Science. 2017. PMID: 28104854 No abstract available.
References
-
- Söding J. Protein homology detection by HMM–HMM comparison. Bioinformatics. 2005;21:951–960. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
