

Spectral consensus strategy for accurate reconstruction of large biological networks
- PMID: 28105915
- PMCID: PMC5249011
- DOI: 10.1186/s12859-016-1308-y
Spectral consensus strategy for accurate reconstruction of large biological networks
Abstract
Background: The last decades witnessed an explosion of large-scale biological datasets whose analyses require the continuous development of innovative algorithms. Many of these high-dimensional datasets are related to large biological networks with few or no experimentally proven interactions. A striking example lies in the recent gut bacterial studies that provided researchers with a plethora of information sources. Despite a deeper knowledge of microbiome composition, inferring bacterial interactions remains a critical step that encounters significant issues, due in particular to high-dimensional settings, unknown gut bacterial taxa and unavoidable noise in sparse datasets. Such data type make any a priori choice of a learning method particularly difficult and urge the need for the development of new scalable approaches.
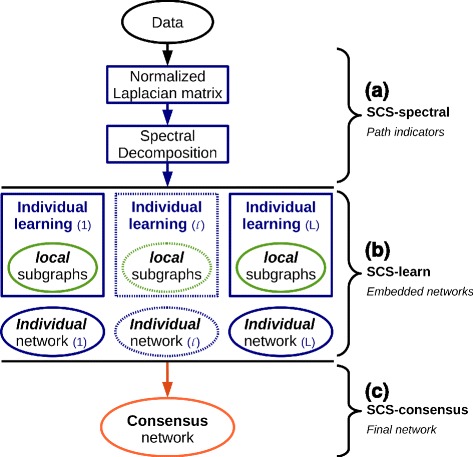
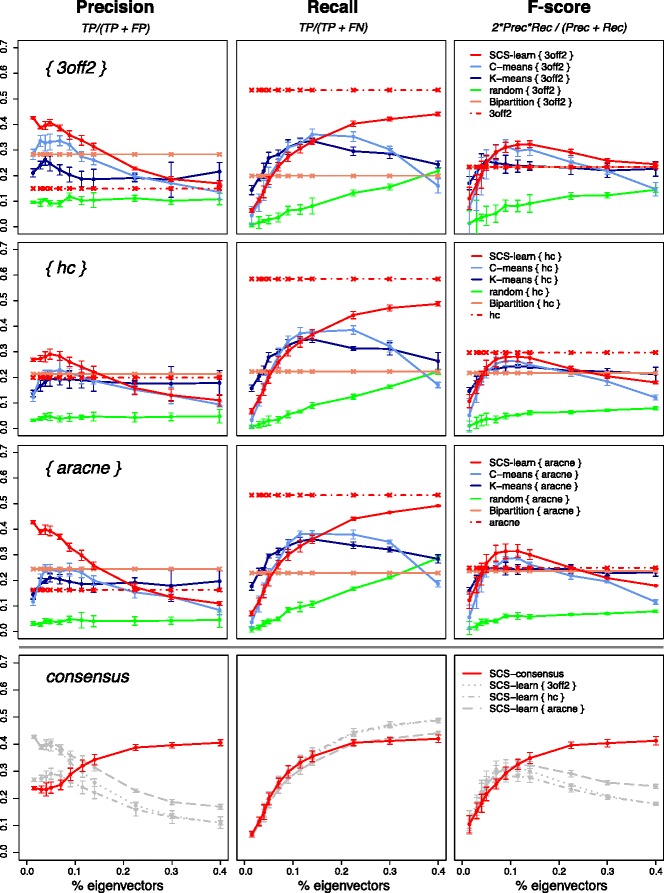
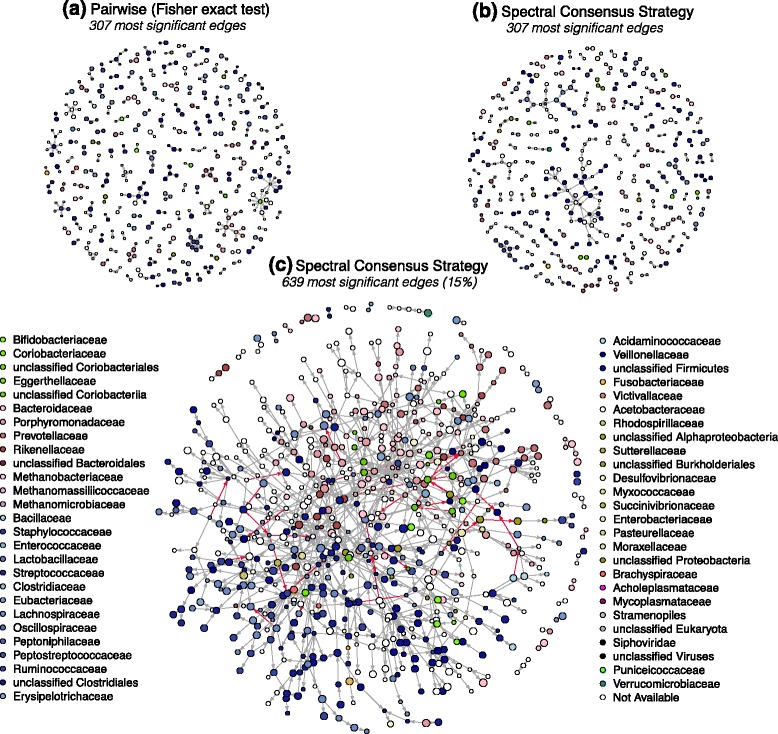
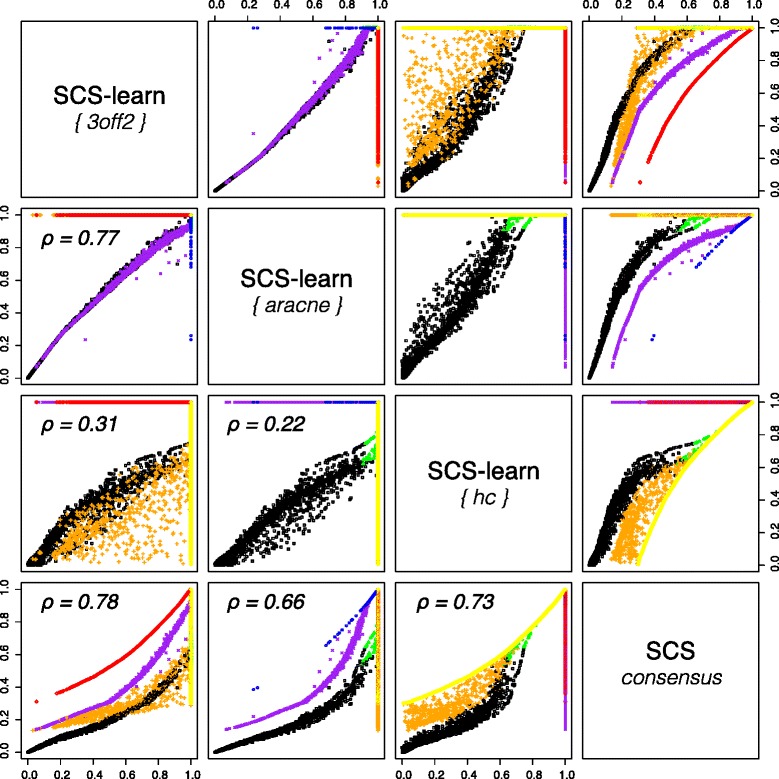
Results: We propose a consensus method based on spectral decomposition, named Spectral Consensus Strategy, to reconstruct large networks from high-dimensional datasets. This novel unsupervised approach can be applied to a broad range of biological networks and the associated spectral framework provides scalability to diverse reconstruction methods. The results obtained on benchmark datasets demonstrate the interest of our approach for high-dimensional cases. As a suitable example, we considered the human gut microbiome co-presence network. For this application, our method successfully retrieves biologically relevant relationships and gives new insights into the topology of this complex ecosystem.
Conclusions: The Spectral Consensus Strategy improves prediction precision and allows scalability of various reconstruction methods to large networks. The integration of multiple reconstruction algorithms turns our approach into a robust learning method. All together, this strategy increases the confidence of predicted interactions from high-dimensional datasets without demanding computations.
Keywords: Community-based method; High-dimensional data; Microbiota; Network reconstruction; Spectral theory.
Figures
References
-
- Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, Kasif S, Collins JJ, Gardner TS. Large-scale mapping and validation of escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5(1):8. doi: 10.1371/journal.pbio.0050008. - DOI - PMC - PubMed
-
- Friedman N, Linial M, Nachman I, Pe’er D. International Conference on Computational Molecular Biology. New York: Mary Ann Liebert, Inc.; 2000. Using Bayesian networks to analyze expression data. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
