

Enzymatic Halogenation and Dehalogenation Reactions: Pervasive and Mechanistically Diverse
- PMID: 28106994
- PMCID: PMC5575885
- DOI: 10.1021/acs.chemrev.6b00571
Enzymatic Halogenation and Dehalogenation Reactions: Pervasive and Mechanistically Diverse
Abstract
Naturally produced halogenated compounds are ubiquitous across all domains of life where they perform a multitude of biological functions and adopt a diversity of chemical structures. Accordingly, a diverse collection of enzyme catalysts to install and remove halogens from organic scaffolds has evolved in nature. Accounting for the different chemical properties of the four halogen atoms (fluorine, chlorine, bromine, and iodine) and the diversity and chemical reactivity of their organic substrates, enzymes performing biosynthetic and degradative halogenation chemistry utilize numerous mechanistic strategies involving oxidation, reduction, and substitution. Biosynthetic halogenation reactions range from simple aromatic substitutions to stereoselective C-H functionalizations on remote carbon centers and can initiate the formation of simple to complex ring structures. Dehalogenating enzymes, on the other hand, are best known for removing halogen atoms from man-made organohalogens, yet also function naturally, albeit rarely, in metabolic pathways. This review details the scope and mechanism of nature's halogenation and dehalogenation enzymatic strategies, highlights gaps in our understanding, and posits where new advances in the field might arise in the near future.
Conflict of interest statement
The authors declare no competing financial interests.
Figures



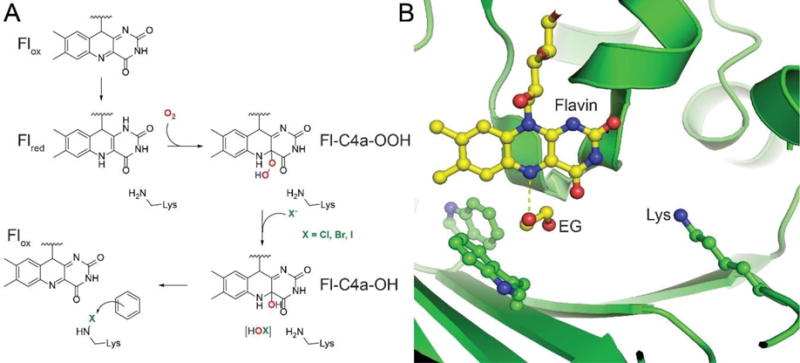






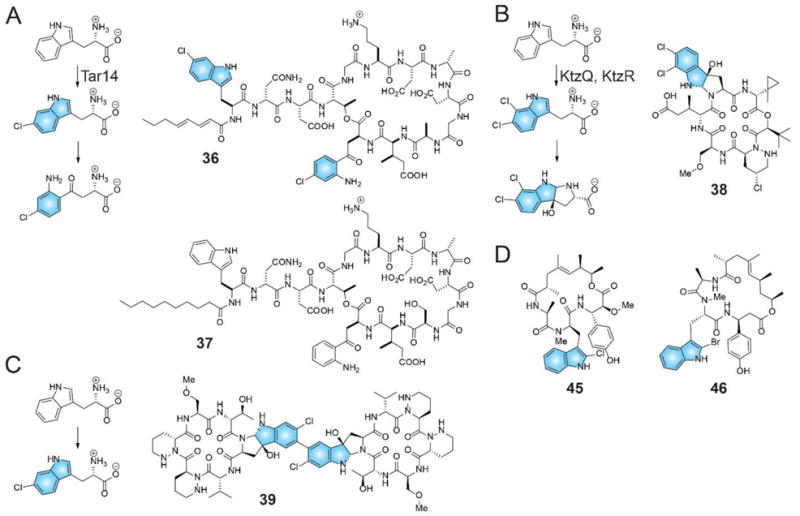




























References
-
- Gribble GW. Naturally occurring organohalogen compounds–a comprehensive survey. Fortschr Chem Org Naturst. 1996;68:1–423. - PubMed
-
- Gribble GW. Naturally Occurring Organohalogen Compounds – A Comprehensive Update. Springer; Vienna: 2010.
-
- Gribble Gw. The natural production of organobromine compounds. Environ Sci Pollut Res Int. 2000;7:37–47. - PubMed
-
- Emerson S, Hedges J. Chemical Oceanography and the Marine Carbon Cycle. 2008
-
- Lohman DC, Edwards DR, Wolfenden R. Catalysis by desolvation: the catalytic prowess of SAM-dependent halide-alkylating enzymes. J Am Chem Soc. 2013;135:14473–14475. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
