

Controlling Tumor Progression with Cyclophosphamide, Vincristine, and Dacarbazine Treatment Improves Survival in Patients with Metastatic and Unresectable Malignant Pheochromocytomas/Paragangliomas
- PMID: 28108930
- PMCID: PMC10355862
- DOI: 10.1007/s12672-017-0284-7
Controlling Tumor Progression with Cyclophosphamide, Vincristine, and Dacarbazine Treatment Improves Survival in Patients with Metastatic and Unresectable Malignant Pheochromocytomas/Paragangliomas
Abstract
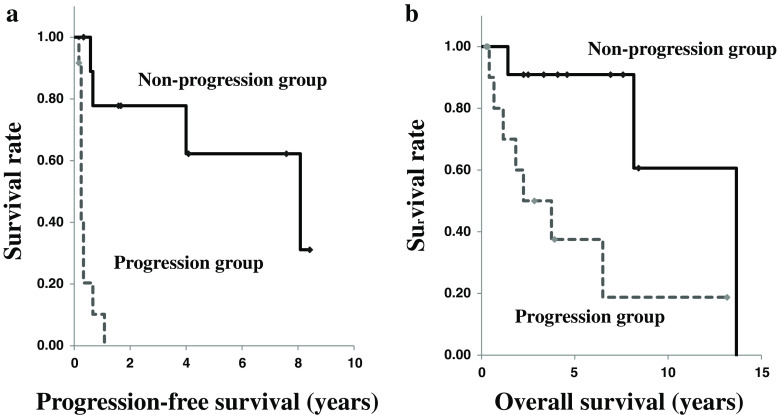
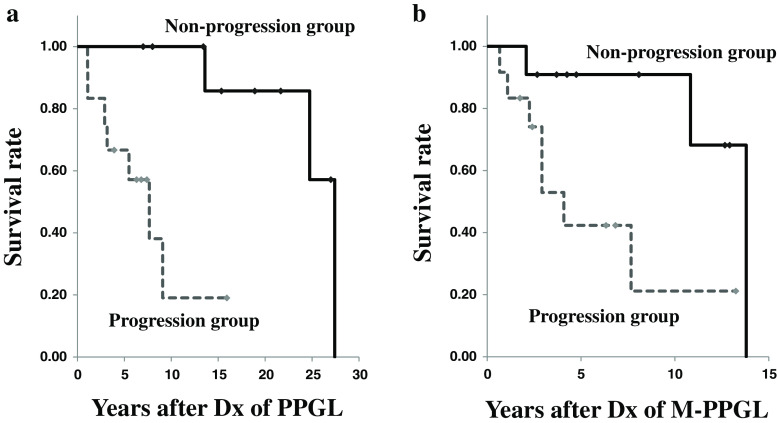
Evidence has not been established to support that combination chemotherapy with cyclophosphamide, vincristine, and dacarbazine (CVD) improves survival in patients with malignant pheochromocytoma and paraganglioma (M-PPGL). To investigate the efficacy of CVD for this disease, we retrospectively analyzed data of 23 patients with metastatic and unresectable M-PPGL (mean age, 41.7 ± 15.4 years) who received at least 2 cycles of this regimen. The follow-up period after initiation of CVD ranged from 0.3 to 13.7 years, with a median of 3.3 years. CVD therapy achieved a complete tumor response (CR) in 1 patient (4%), a partial response (PR) in 5 (22%), stable disease (SD) in 5 (22%), and progressive disease (PD) in 13 (52%), respectively. All of the responders (CR and PR) but 6% of the non-responders (SD and PD) showed substantial biochemical improvement. The progression-free survival period in the responders was significantly longer than in the non-responders (p < 0.01). Although the overall survival and survival after the diagnosis of M-PPGL were longer in the responders than the non-responders, the difference was not statistically significant (p = 0.08). The progression-free and overall survival period were significantly longer in the non-progression group (CR, PR, and SD) than in the progression group (PD) (1.7 ± 3.3 vs. 0.3 ± 0.3 years, p < 0.01, and 4.6 ± 3.6 vs. 2.0 ± 3.7 years, p = 0.01, respectively). It is therefore suggested that CVD chemotherapy could be useful in controlling tumor progression and improving survival in patients with metastatic and progressive M-PPGL.
Keywords: Antineoplastic combined chemotherapy; CVD therapy; Malignant pheochromocytoma; Myelodysplastic syndromes; Paraganglioma; Survival time.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
References
-
- Eisenhofer G, Bornstein SR, Brouwers FM, Cheung NK, Dahia PL, de Krijger RR, Giordano TJ, Greene LA, Goldstein DS, Lehnert H, Manger WM, Maris JM, Neumann HP, Pacak K, Shulkin BL, Smith DI, Tischler AS, Young WF., Jr Malignant pheochromocytoma: current status and initiatives for future progress. Endocr Relat Cancer. 2004;11:423–436. doi: 10.1677/erc.1.00829. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
