

Ligand and Target Discovery by Fragment-Based Screening in Human Cells
- PMID: 28111073
- PMCID: PMC5632530
- DOI: 10.1016/j.cell.2016.12.029
Ligand and Target Discovery by Fragment-Based Screening in Human Cells
Abstract
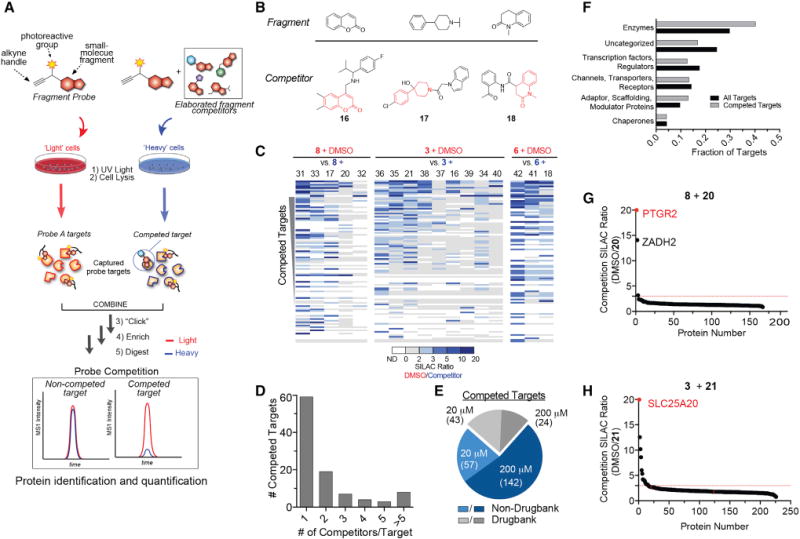
Advances in the synthesis and screening of small-molecule libraries have accelerated the discovery of chemical probes for studying biological processes. Still, only a small fraction of the human proteome has chemical ligands. Here, we describe a platform that marries fragment-based ligand discovery with quantitative chemical proteomics to map thousands of reversible small molecule-protein interactions directly in human cells, many of which can be site-specifically determined. We show that fragment hits can be advanced to furnish selective ligands that affect the activity of proteins heretofore lacking chemical probes. We further combine fragment-based chemical proteomics with phenotypic screening to identify small molecules that promote adipocyte differentiation by engaging the poorly characterized membrane protein PGRMC2. Fragment-based screening in human cells thus provides an extensive proteome-wide map of protein ligandability and facilitates the coordinated discovery of bioactive small molecules and their molecular targets.
Keywords: FBLD; PGRMC2; adipogenesis; chemical probes; chemical proteomics; fragment-based ligand discovery; ligands; mass spectrometry; phenotypic screening; photoreactivity.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures






Comment in
-
Ligandable proteome mapping.Nat Methods. 2019 Dec;16(12):1204. doi: 10.1038/s41592-019-0673-7. Nat Methods. 2019. PMID: 31780831 No abstract available.
References
-
- Bembenek SD, Tounge BA, Reynolds CH. Ligand efficiency and fragment-based drug discovery. Drug Discov. Today. 2009;14:278–283. - PubMed
-
- Cahill MA. Progesterone receptor membrane component 1: An integrative review. J Steroid Biochem Mol Biol. 2007;105:16–36. - PubMed
-
- Chen JJ, London IM. Hemin enhances the differentiation of mouse 3T3 cells to adipocytes. Cell. 1981;26:117–122. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
