

ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion
- PMID: 28111074
- PMCID: PMC5310835
- DOI: 10.1016/j.cell.2016.12.044
ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion
Abstract
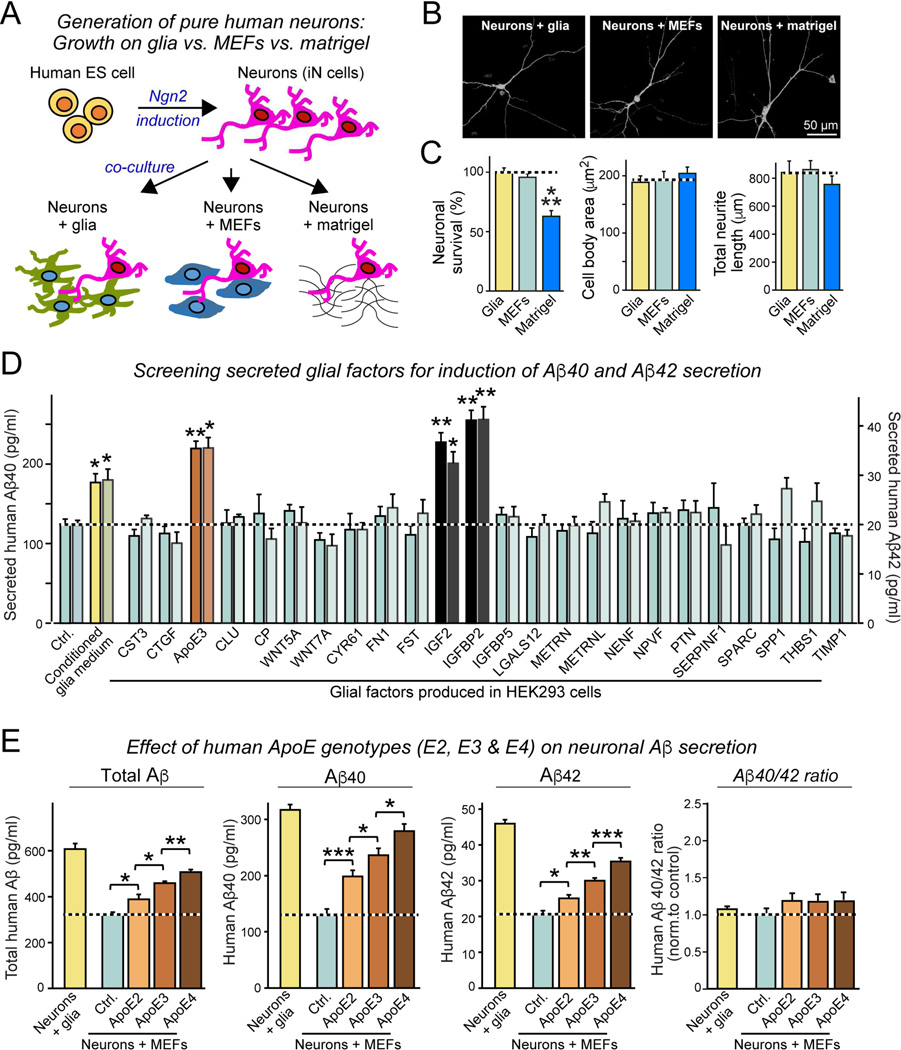
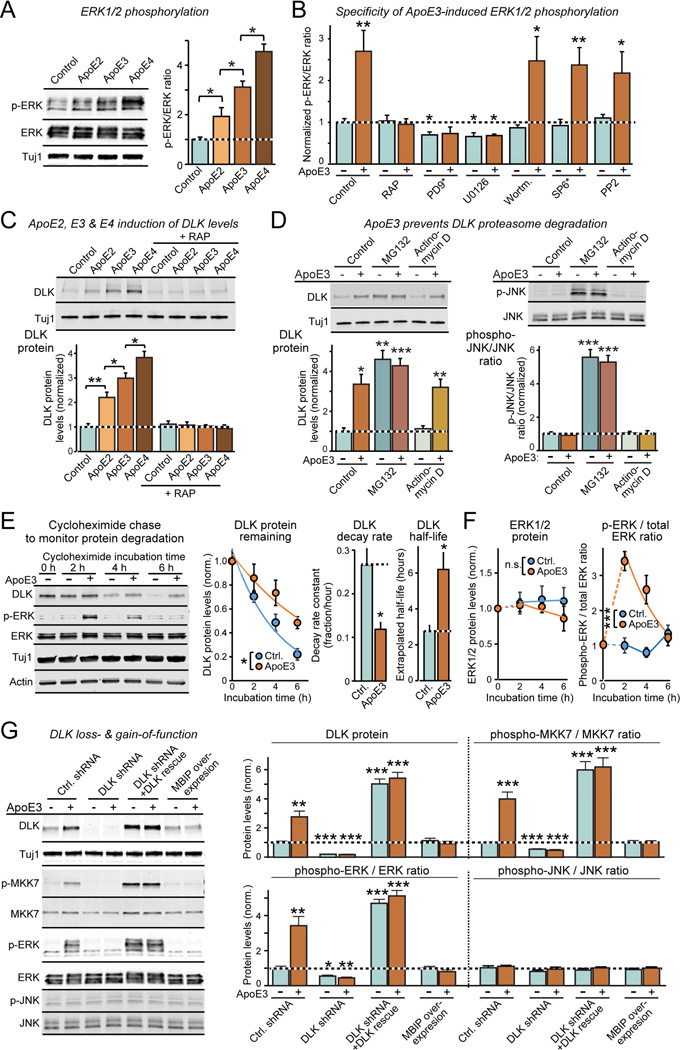
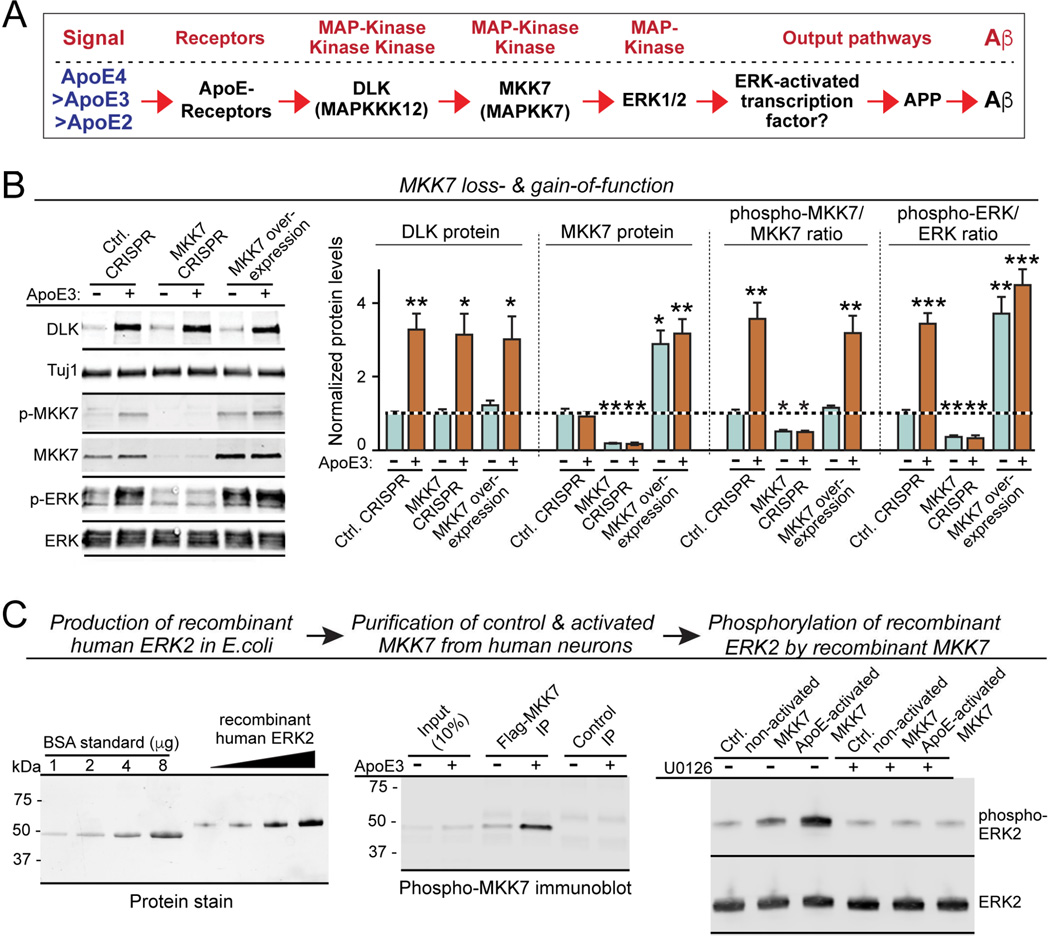
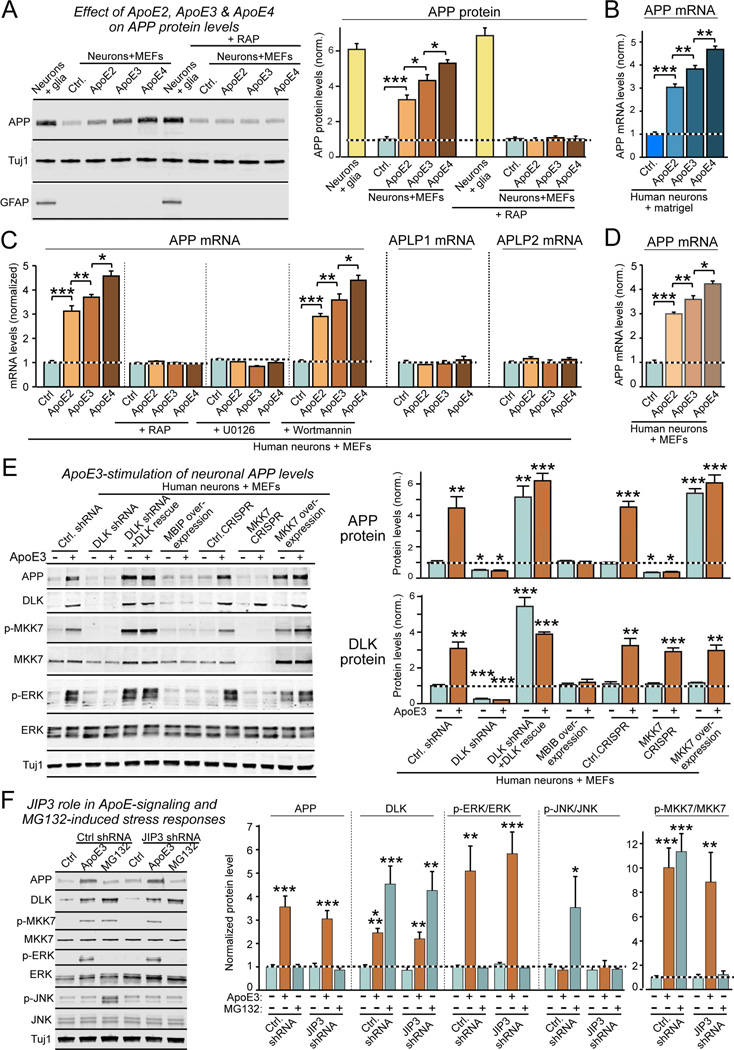
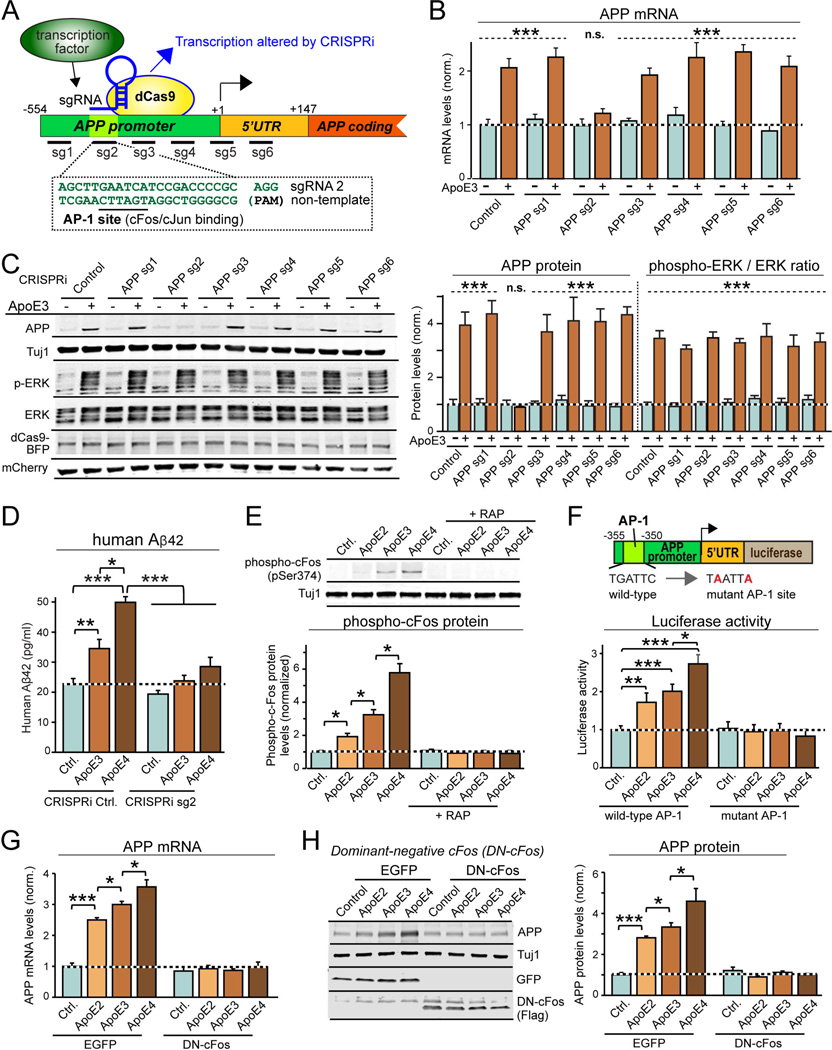
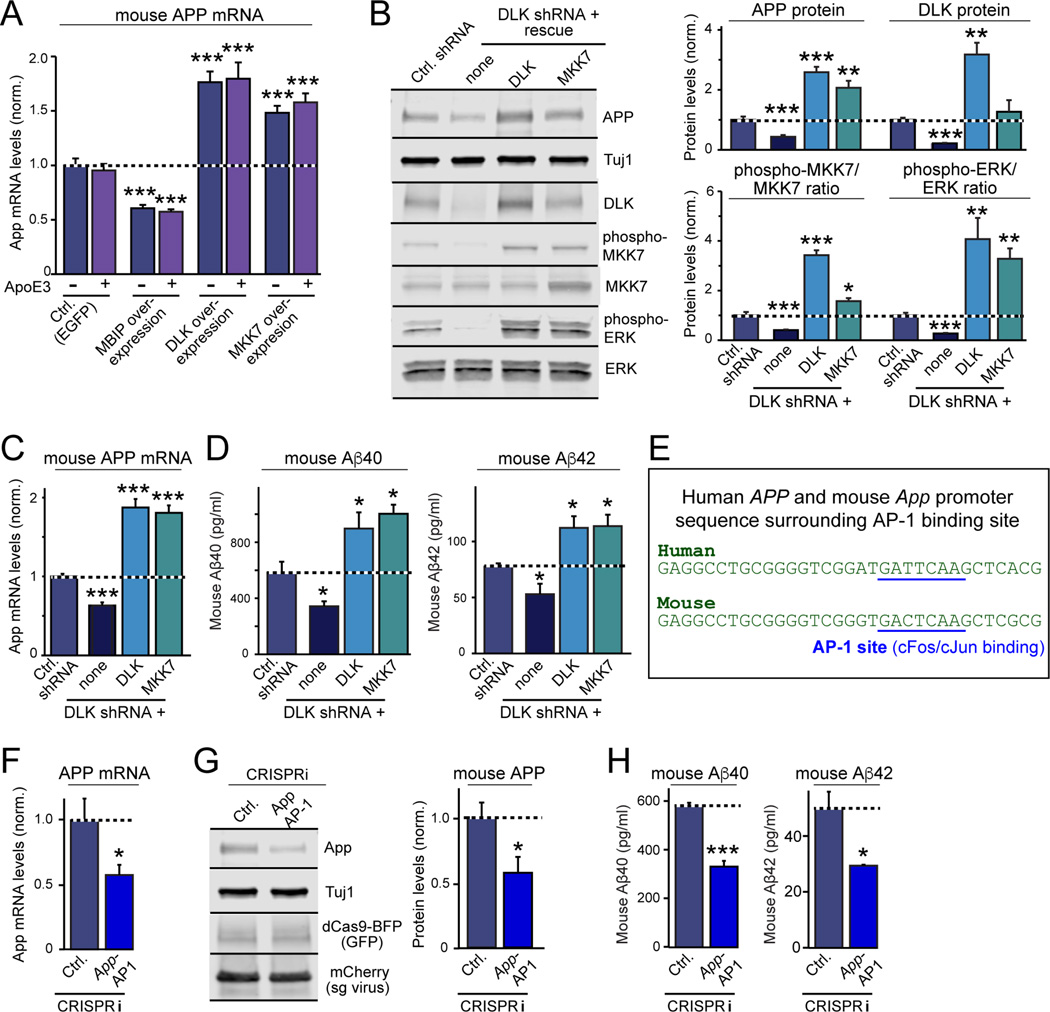
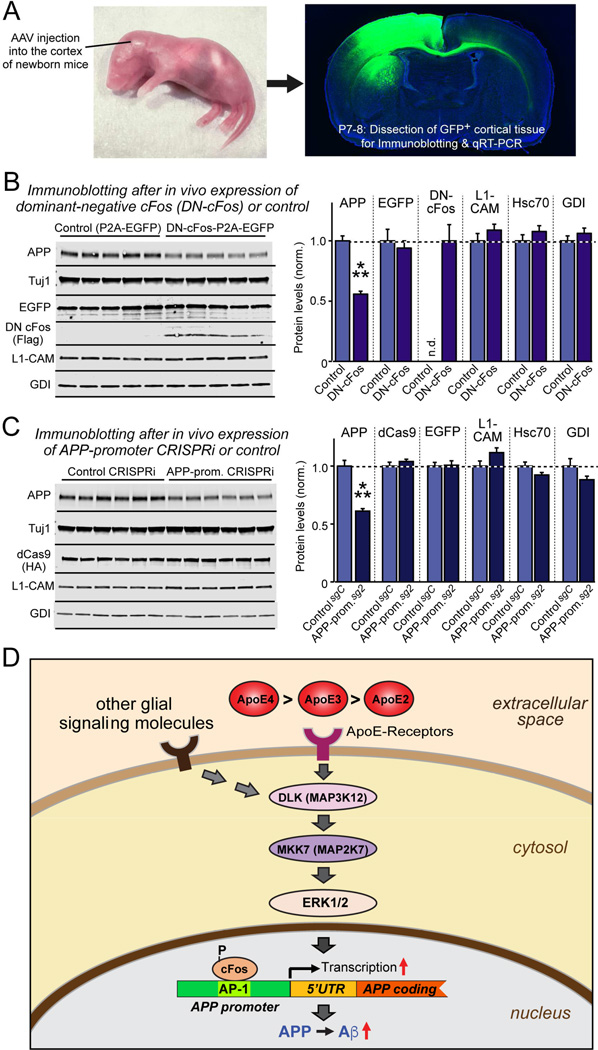
Human apolipoprotein E (ApoE) apolipoprotein is primarily expressed in three isoforms (ApoE2, ApoE3, and ApoE4) that differ only by two residues. ApoE4 constitutes the most important genetic risk factor for Alzheimer's disease (AD), ApoE3 is neutral, and ApoE2 is protective. How ApoE isoforms influence AD pathogenesis, however, remains unclear. Using ES-cell-derived human neurons, we show that ApoE secreted by glia stimulates neuronal Aβ production with an ApoE4 > ApoE3 > ApoE2 potency rank order. We demonstrate that ApoE binding to ApoE receptors activates dual leucine-zipper kinase (DLK), a MAP-kinase kinase kinase that then activates MKK7 and ERK1/2 MAP kinases. Activated ERK1/2 induces cFos phosphorylation, stimulating the transcription factor AP-1, which in turn enhances transcription of amyloid-β precursor protein (APP) and thereby increases amyloid-β levels. This molecular mechanism also regulates APP transcription in mice in vivo. Our data describe a novel signal transduction pathway in neurons whereby ApoE activates a non-canonical MAP kinase cascade that enhances APP transcription and amyloid-β synthesis.
Keywords: APP; Alzheimer’s disease; ApoE; Aβ; CRISPR; CRISPRi; DLK; MAP kinase signaling; amyloid precursor protein; apolipoprotein E; beta amyloid; cFos; dual leucine-zipper kinase; transcription.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Brouwers N, Sleegers K, Engelborghs S, Bogaerts V, Serneels S, Kamali K, Corsmit E, De Leenheir E, Martin JJ, De Deyn PP, et al. Genetic risk and transcriptional variability of amyloid precursor protein in Alzheimer’s disease. Brain. 2006;129:2984–2991. - PubMed
-
- Christ A, Christa A, Kur E, Lioubinski O, Bachmann S, Willnow TE, Hammes A. LRP2 is an auxiliary SHH receptor required to condition the forebrain ventral midline for inductive signals. Dev. Cell. 2012;22:268–278. - PubMed
-
- De Strooper B, Karran E. The cellular phase of Alzheimer’s disease. Cell. 2016;164:603–615. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
