

A tale of two glutaminases: homologous enzymes with distinct roles in tumorigenesis
- PMID: 28111979
- PMCID: PMC5558546
- DOI: 10.4155/fmc-2016-0190
A tale of two glutaminases: homologous enzymes with distinct roles in tumorigenesis
Erratum in
-
Corrigendum.Future Med Chem. 2017 Apr;9(5):527. doi: 10.4155/fmc-2016-0190c1. Epub 2017 Mar 23. Future Med Chem. 2017. PMID: 28332860 Free PMC article. No abstract available.
Abstract
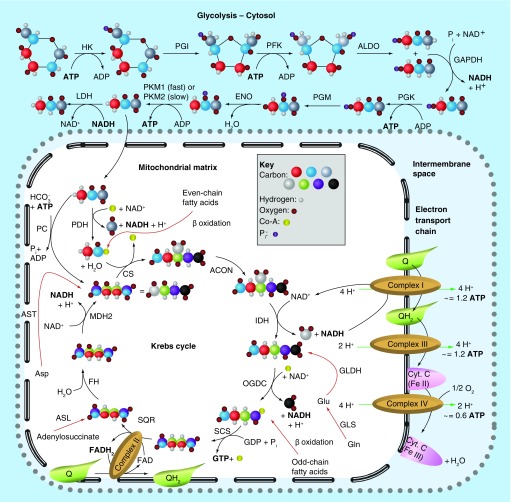
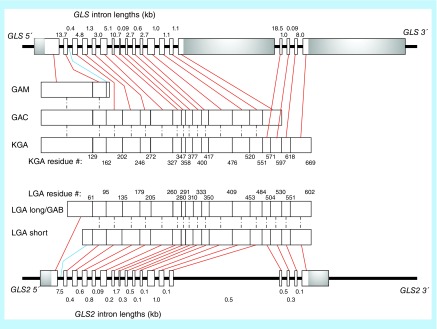
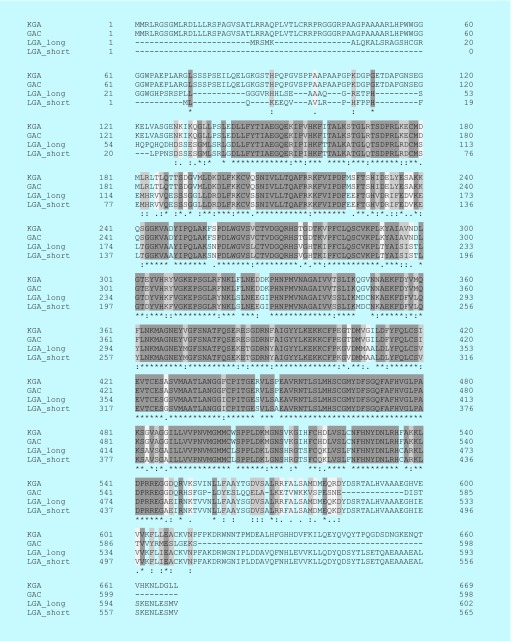
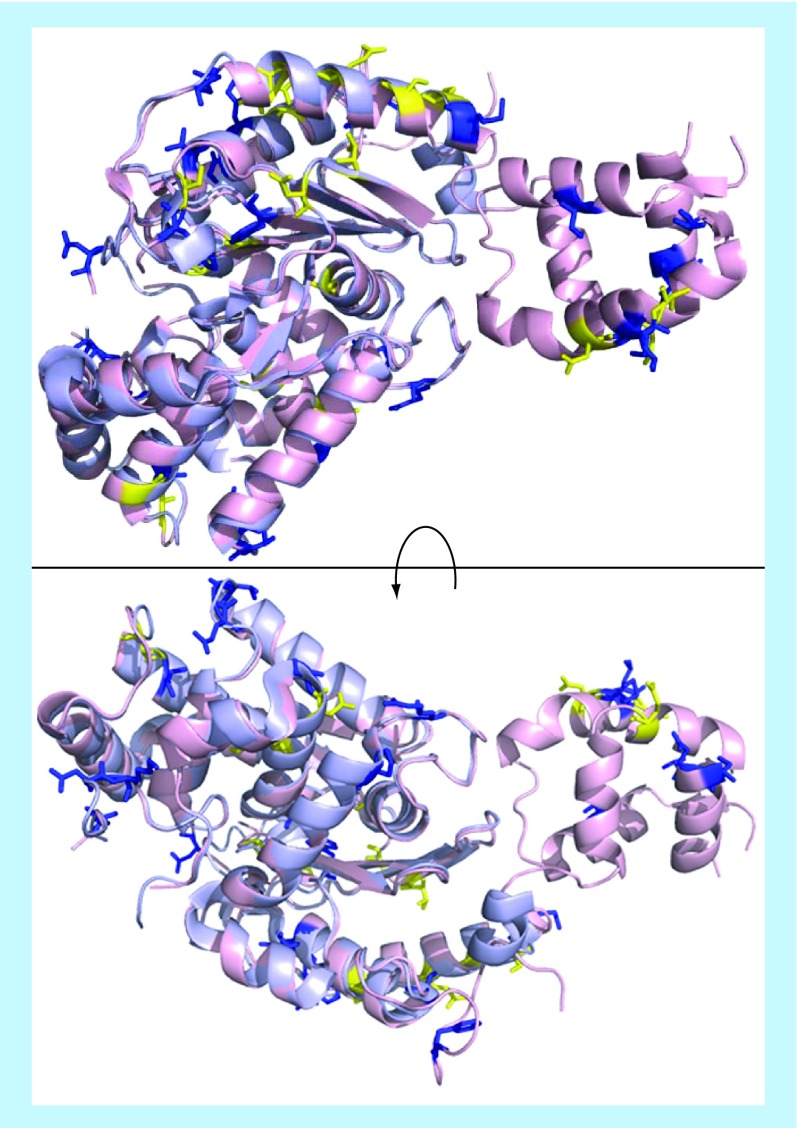
Many cancer cells exhibit an altered metabolic phenotype, in which glutamine consumption is upregulated relative to healthy cells. This metabolic reprogramming often depends upon mitochondrial glutaminase activity, which converts glutamine to glutamate, a key precursor for biosynthetic and bioenergetic processes. Two isozymes of glutaminase exist, a kidney-type (GLS) and a liver-type enzyme (GLS2 or LGA). While a majority of studies have focused on GLS, here we summarize key findings on both glutaminases, describing their structure and function, their roles in cancer and pharmacological approaches to inhibiting their activities.
Keywords: 968; BPTES; CB-839; Warburg effect; cancer; glutaminase; inhibitor; metabolism.
Conflict of interest statement
This work was supported by funding from the NIH (GM040654, GM047458, CA201402). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Figures
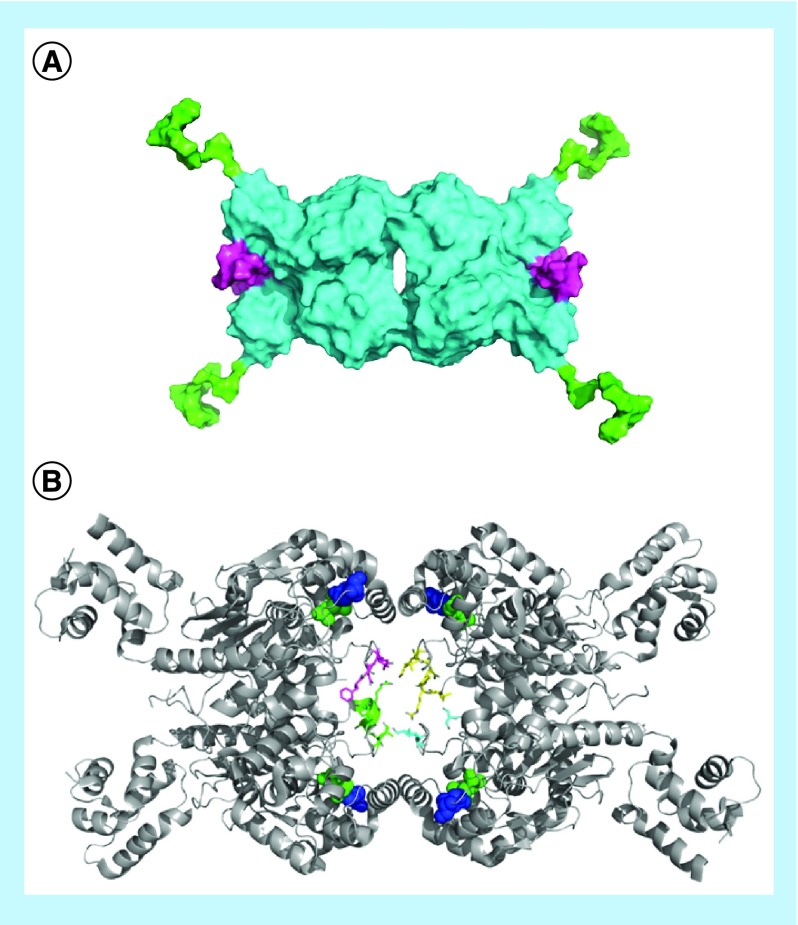
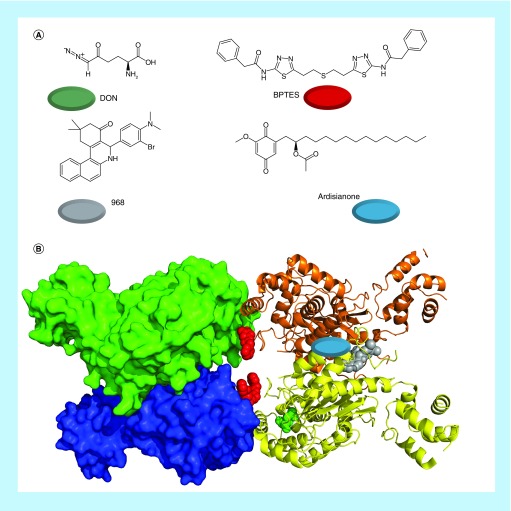
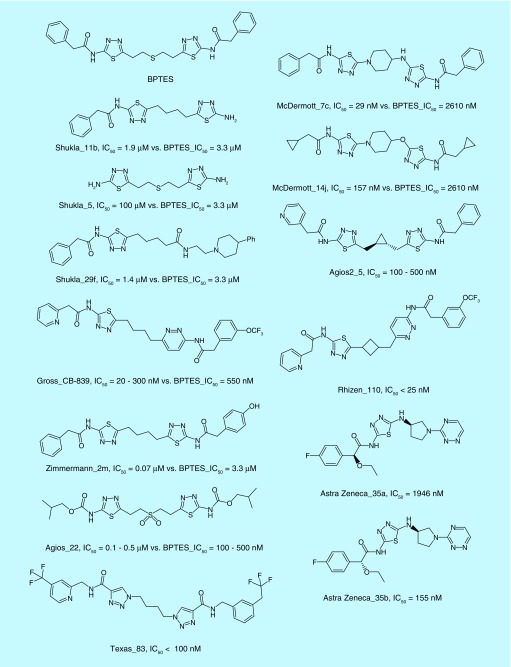
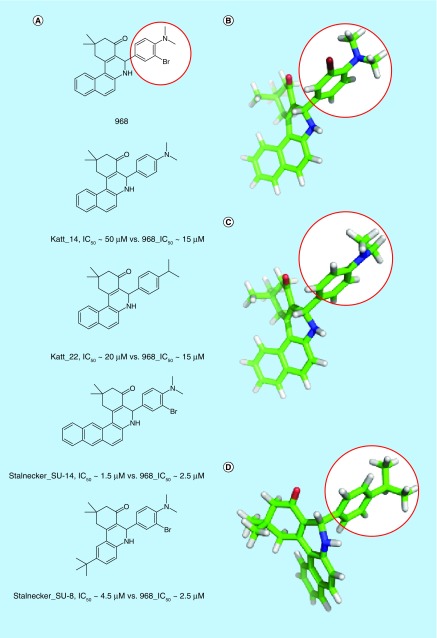
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous