

BX-795 inhibits HSV-1 and HSV-2 replication by blocking the JNK/p38 pathways without interfering with PDK1 activity in host cells
- PMID: 28112176
- PMCID: PMC5342671
- DOI: 10.1038/aps.2016.160
BX-795 inhibits HSV-1 and HSV-2 replication by blocking the JNK/p38 pathways without interfering with PDK1 activity in host cells
Abstract
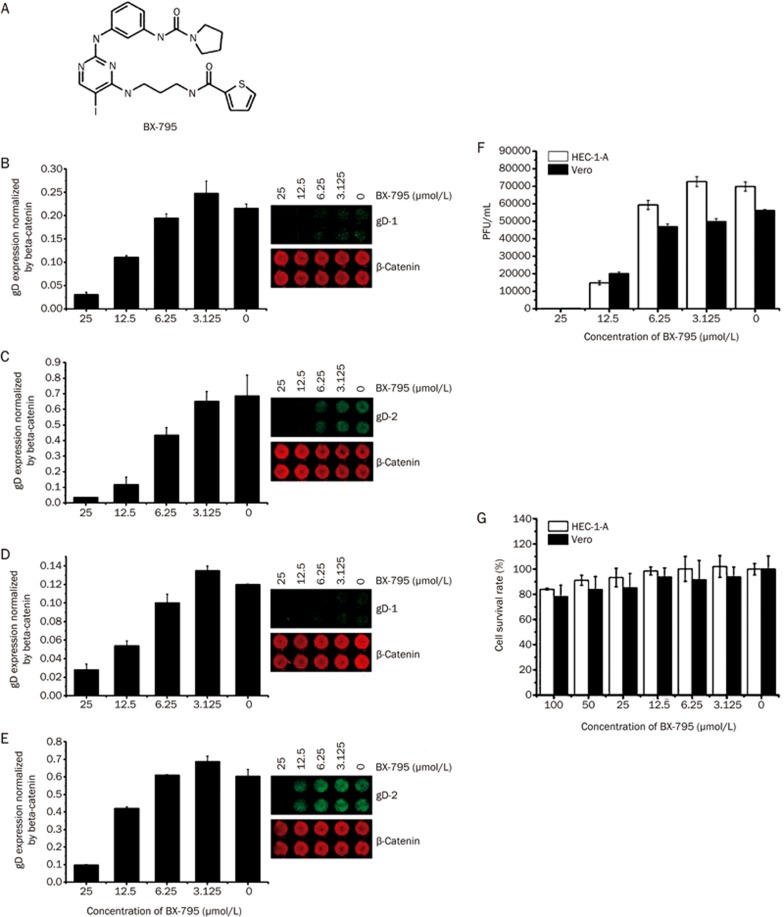
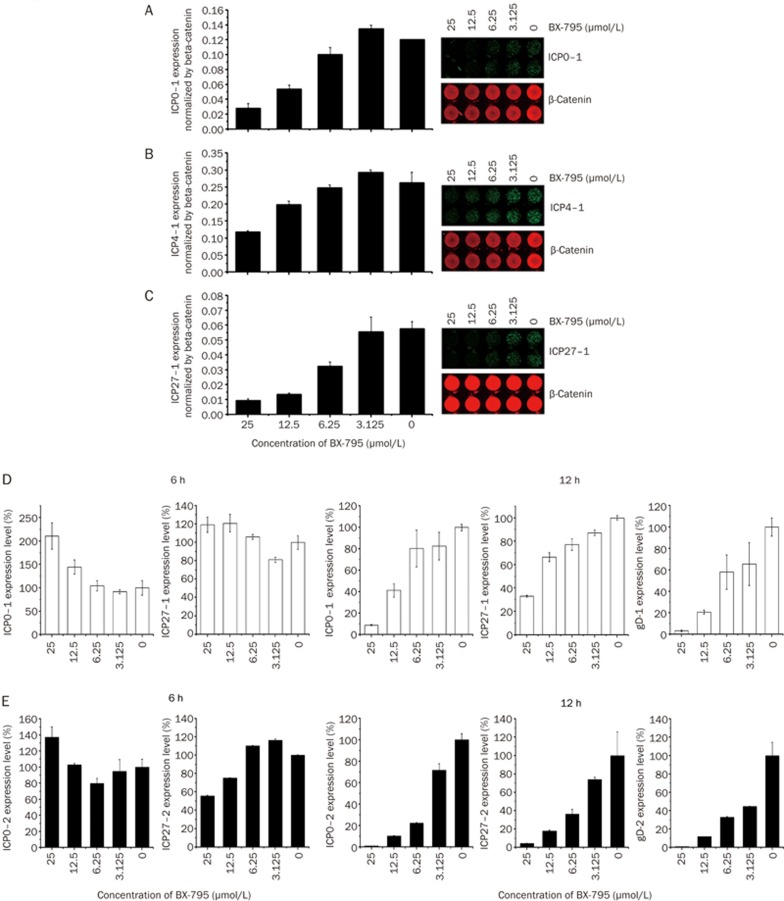
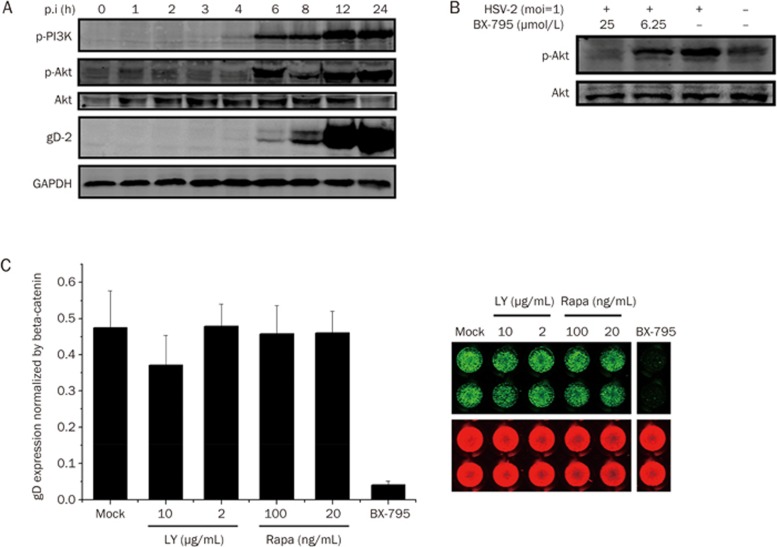
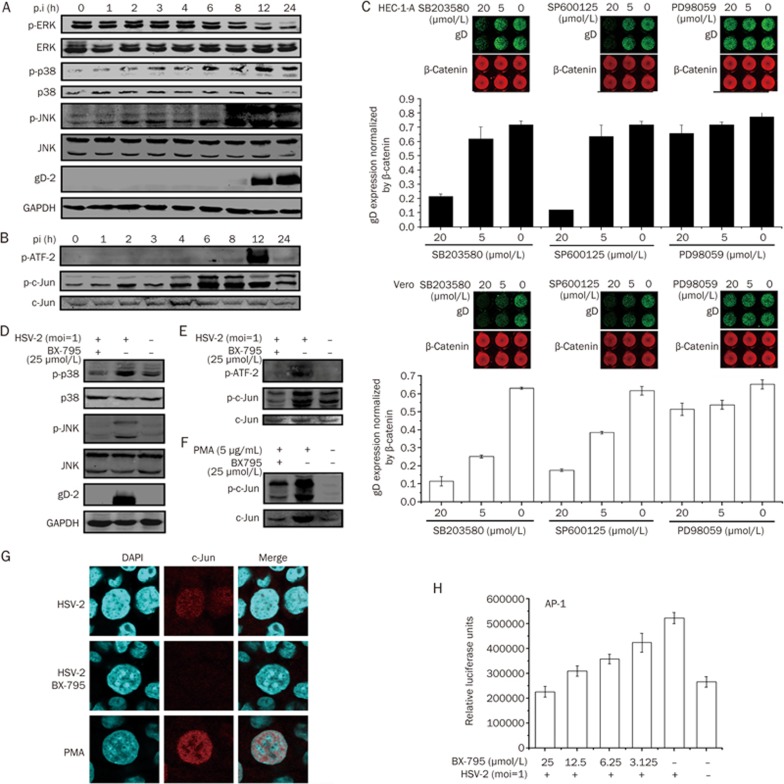
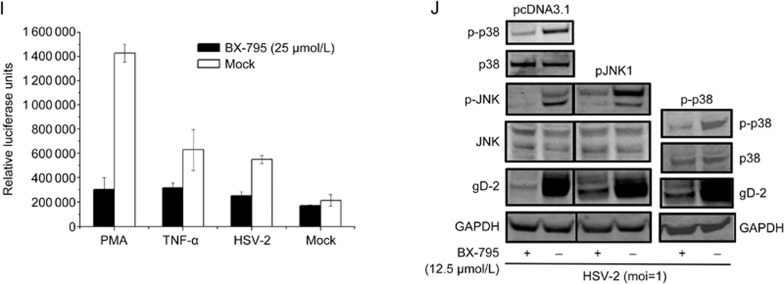
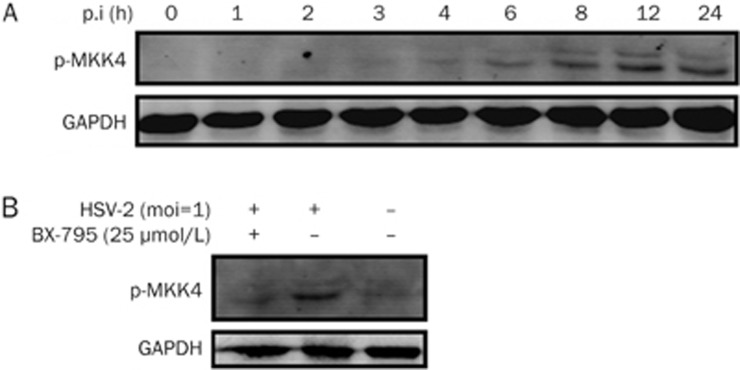
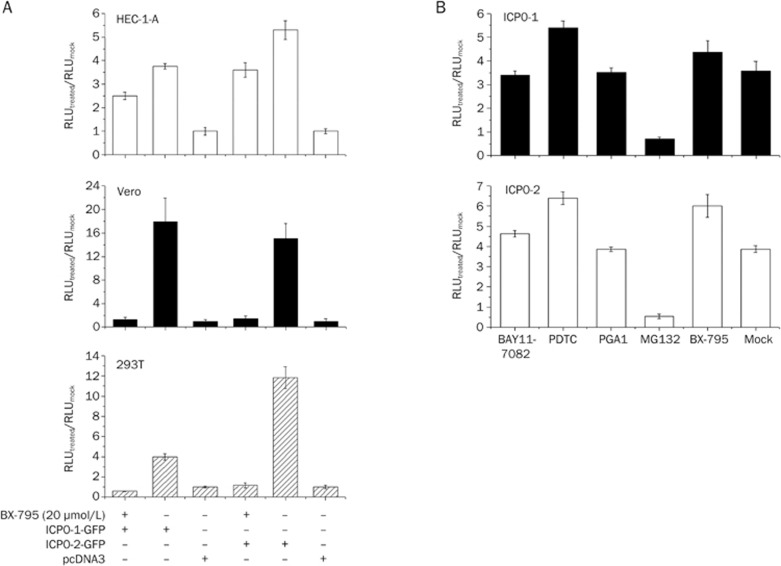
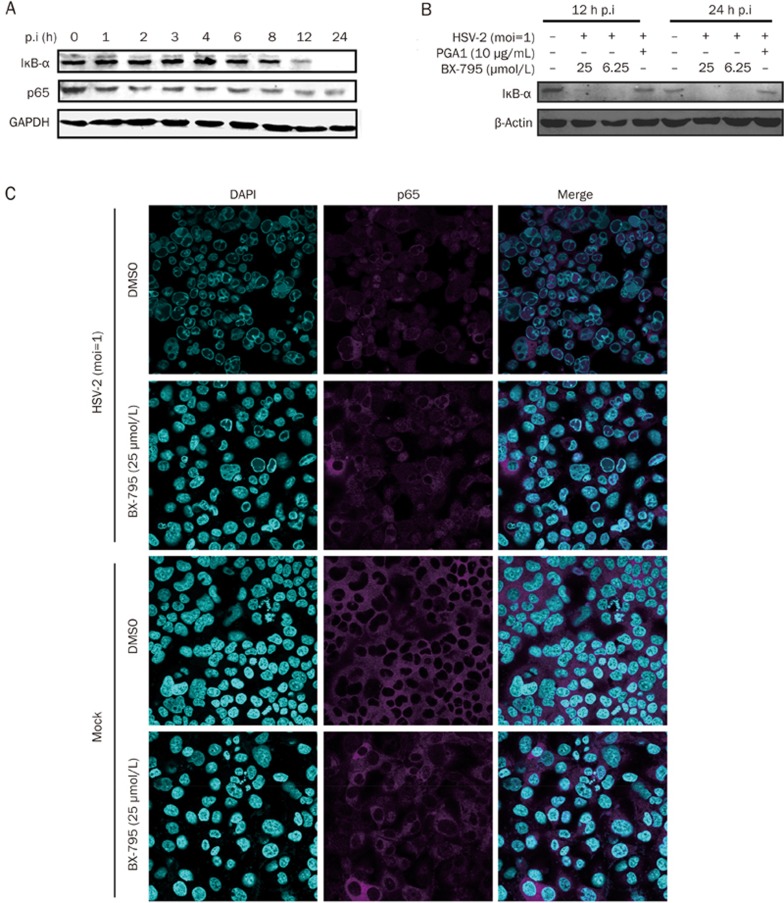
BX-795 is an inhibitor of 3-phosphoinositide-dependent kinase 1 (PDK1), but also a potent inhibitor of the IKK-related kinase, TANKbinding kinase 1 (TBK1) and IKKɛ. In this study we attempted to elucidate the molecular mechanism(s) underlying the inhibition of BX-795 on Herpes simplex virus (HSV) replication. HEC-1-A or Vero cells were treated with BX-795 and infected with HSV-1 or HSV-2 for different periods. BX-795 (3.125-25 μmol/L) dose-dependently suppressed HSV-2 replication, and displayed a low cytotoxicity to the host cells. BX-795 treatment dose-dependently suppressed the expression of two HSV immediate-early (IE) genes (ICP0 and ICP27) and the late gene (gD) at 12 h postinfection. HSV-2 infection resulted in the activation of PI3K and Akt in the host cells, and BX-795 treatment inhibited HSV-2-induced Akt phosphorylation and activation. However, the blockage of PI3K/Akt/mTOR with LY294002 and rapamycin did not affect HSV-2 replication. HSV-2 infection increased the phosphorylation of JNK and p38, and reduced ERK phosphorylation at 8 h postinfection in the host cells; BX-795 treatment inhibited HSV-2-induced activation of JNK and p38 MAP kinase as well as the phosphorylation of c-Jun and ATF-2, the downstream targets of JNK and p38 MAP kinase. Furthermore, SB203580 (a p38 inhibitor) or SP600125 (a JNK inhibitor) dose-dependently inhibited the viral replication in the host cells, whereas PD98059 (an ERK inhibitor) was not effective. Moreover, BX-795 blocked PMA-stimulated c-Jun activation as well as HSV-2-mediated c-Jun nuclear translocation. BX-795 dose-dependently inhibited HSV-2, PMA, TNF-α-stimulated AP-1 activation, but not HSV-induced NF-κB activation. Overexpression of p38/JNK attenuated the inhibitory effect of BX-795 on HSV replication. BX-795 completely blocked HSV-2-induced MKK4 phosphorylation, suggesting that BX-795 acting upstream of JNK and p38 MAP kinase. In conclusion, this study identifies the anti-HSV activity of BX-795 and its targeting of the JNK/p38 MAP kinase pathways in host cells.
Figures
Similar articles
-
Resveratrol suppresses nuclear factor-kappaB in herpes simplex virus infected cells.Antiviral Res. 2006 Dec;72(3):242-51. doi: 10.1016/j.antiviral.2006.06.011. Epub 2006 Jul 14. Antiviral Res. 2006. PMID: 16876885
-
Boswellia serrata oleo-gum-resin and β-boswellic acid inhibits HSV-1 infection in vitro through modulation of NF-кB and p38 MAP kinase signaling.Phytomedicine. 2018 Dec 1;51:94-103. doi: 10.1016/j.phymed.2018.10.016. Epub 2018 Oct 16. Phytomedicine. 2018. PMID: 30466633
-
Opposite Roles of RNase and Kinase Activities of Inositol-Requiring Enzyme 1 (IRE1) on HSV-1 Replication.Viruses. 2017 Aug 23;9(9):235. doi: 10.3390/v9090235. Viruses. 2017. PMID: 28832521 Free PMC article.
-
p38 activation and viral infection.Expert Rev Mol Med. 2022 Jan 21;24:e4. doi: 10.1017/erm.2021.29. Expert Rev Mol Med. 2022. PMID: 35060846 Review.
-
New insights on thyroid hormone mediated regulation of herpesvirus infections.Cell Biosci. 2017 Mar 21;7:13. doi: 10.1186/s13578-017-0140-z. eCollection 2017. Cell Biosci. 2017. PMID: 28344765 Free PMC article. Review.
Cited by
-
A new advanced in silico drug discovery method for novel coronavirus (SARS-CoV-2) with tensor decomposition-based unsupervised feature extraction.PLoS One. 2020 Sep 11;15(9):e0238907. doi: 10.1371/journal.pone.0238907. eCollection 2020. PLoS One. 2020. PMID: 32915876 Free PMC article.
-
Antiviral effect of Chinese herbal prescription JieZe-1 on adhesion and penetration of VK2/E6E7 with herpes simplex viruses type 2.J Ethnopharmacol. 2020 Mar 1;249:112405. doi: 10.1016/j.jep.2019.112405. Epub 2019 Nov 16. J Ethnopharmacol. 2020. PMID: 31743766 Free PMC article.
-
Unique Attributes of Guinea Pigs as New Models to Study Ocular Herpes Pathophysiology and Recurrence.Invest Ophthalmol Vis Sci. 2023 Nov 1;64(14):41. doi: 10.1167/iovs.64.14.41. Invest Ophthalmol Vis Sci. 2023. PMID: 38015175 Free PMC article.
-
Gasdermin D aggravates a mouse model of radiation-induced liver disease by promoting chemokine secretion and neutrophil recruitment.Nat Commun. 2025 Jul 2;16(1):6064. doi: 10.1038/s41467-025-61397-7. Nat Commun. 2025. PMID: 40603284 Free PMC article.
-
The roles of signaling pathways in SARS-CoV-2 infection; lessons learned from SARS-CoV and MERS-CoV.Arch Virol. 2021 Mar;166(3):675-696. doi: 10.1007/s00705-021-04958-7. Epub 2021 Jan 18. Arch Virol. 2021. PMID: 33462671 Free PMC article. Review.
References
-
- Freeman EE, Weiss HA, Glynn JR, Cross PL, Whitworth JA, Hayes RJ. Herpes simplex virus 2 infection increases HIV acquisition in men and women: systematic review and meta-analysis of longitudinal studies. AIDS 2006; 20: 73. - PubMed
-
- Morfin F, Thouvenot D. Herpes simplex virus resistance to antiviral drugs. J Clin Virol 2003; 26: 29–37. - PubMed
-
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 2001; 81: 807–69. - PubMed
-
- Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol 2002; 4: E131–6. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
