

Metabolic shift in the emergence of hyperinvasive pandemic meningococcal lineages
- PMID: 28112239
- PMCID: PMC5282872
- DOI: 10.1038/srep41126
Metabolic shift in the emergence of hyperinvasive pandemic meningococcal lineages
Abstract
Hyperinvasive lineages of Neisseria meningitidis, which persist despite extensive horizontal genetic exchange, are a major cause of meningitis and septicaemia worldwide. Over the past 50 years one such lineage of meningococci, known as serogroup A, clonal complex 5 (A:cc5), has caused three successive pandemics, including epidemics in sub-Saharan Africa. Although the principal antigens that invoke effective immunity have remained unchanged, distinct A:cc5 epidemic clones have nevertheless emerged. An analysis of whole genome sequence diversity among 153 A:cc5 isolates identified eleven genetic introgression events in the emergence of the epidemic clones, which primarily involved variants of core genes encoding metabolic processes. The acquired DNA was identical to that found over many years in other, unrelated, hyperinvasive meningococci, suggesting that the epidemic clones emerged by acquisition of pre-existing metabolic gene variants, rather than 'virulence' associated or antigen-encoding genes. This is consistent with mathematical models which predict the association of transmission fitness with the emergence and maintenance of virulence in recombining commensal organisms.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
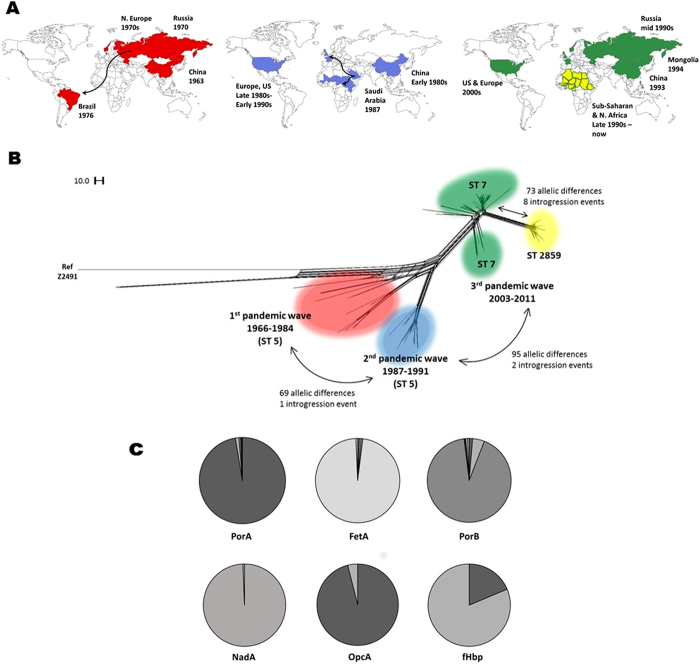
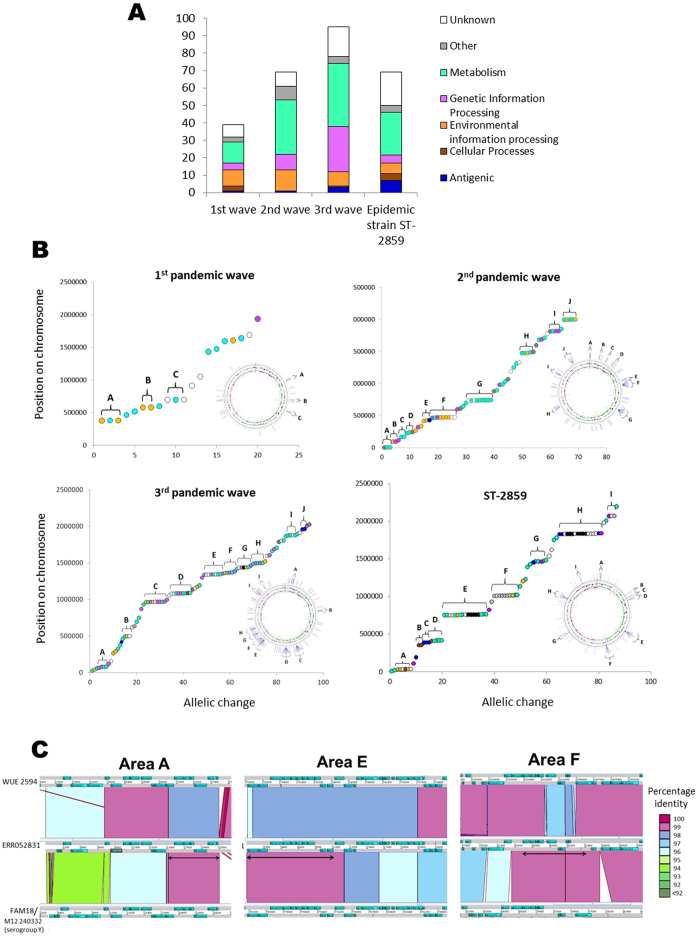
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
