

A comprehensive global genotype-phenotype database for rare diseases
- PMID: 28116331
- PMCID: PMC5241210
- DOI: 10.1002/mgg3.262
A comprehensive global genotype-phenotype database for rare diseases
Abstract
Background: The ability to discover genetic variants in a patient runs far ahead of the ability to interpret them. Databases with accurate descriptions of the causal relationship between the variants and the phenotype are valuable since these are critical tools in clinical genetic diagnostics. Here, we introduce a comprehensive and global genotype-phenotype database focusing on rare diseases.
Methods: This database (CentoMD ®) is a browser-based tool that enables access to a comprehensive, independently curated system utilizing stringent high-quality criteria and a quickly growing repository of genetic and human phenotype ontology (HPO)-based clinical information. Its main goals are to aid the evaluation of genetic variants, to enhance the validity of the genetic analytical workflow, to increase the quality of genetic diagnoses, and to improve evaluation of treatment options for patients with hereditary diseases. The database software correlates clinical information from consented patients and probands of different geographical backgrounds with a large dataset of genetic variants and, when available, biomarker information. An automated follow-up tool is incorporated that informs all users whenever a variant classification has changed. These unique features fully embedded in a CLIA/CAP-accredited quality management system allow appropriate data quality and enhanced patient safety.
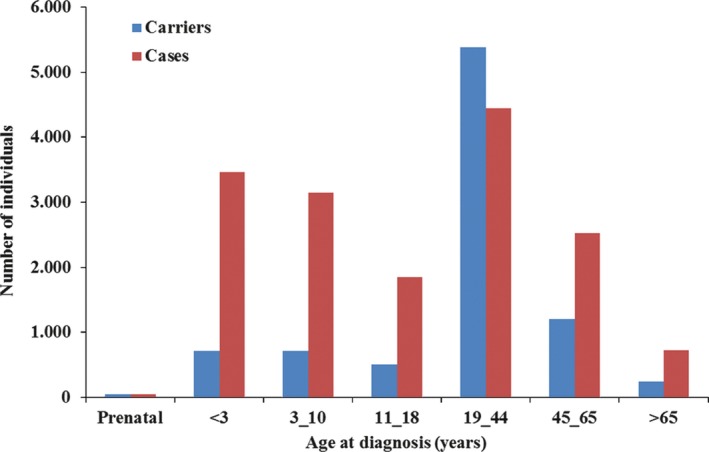
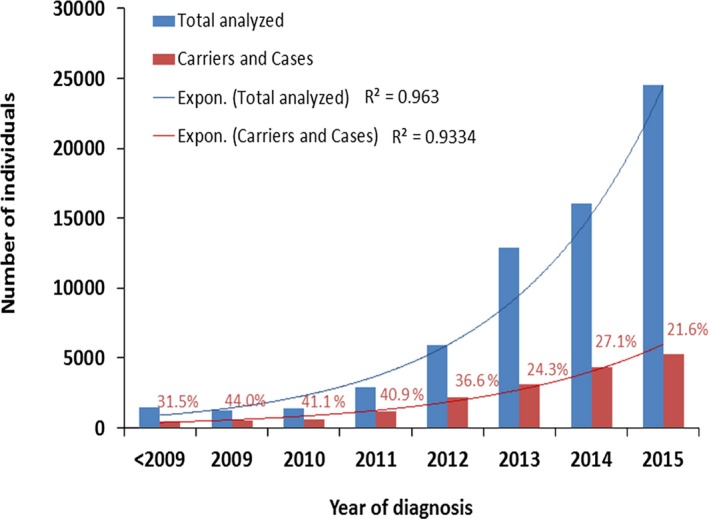
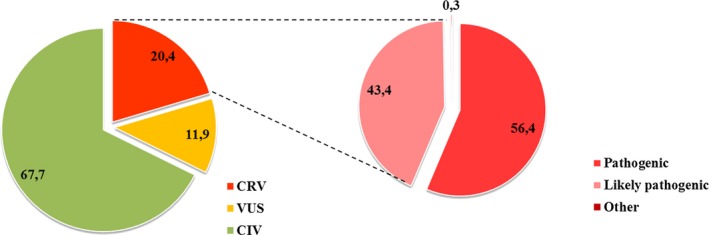
Results: More than 100,000 genetically screened individuals are documented in the database, resulting in more than 470 million variant detections. Approximately, 57% of the clinically relevant and uncertain variants in the database are novel. Notably, 3% of the genetic variants identified and previously reported in the literature as being associated with a particular rare disease were reclassified, based on internal evidence, as clinically irrelevant.
Conclusions: The database offers a comprehensive summary of the clinical validity and causality of detected gene variants with their associated phenotypes, and is a valuable tool for identifying new disease genes through the correlation of novel genetic variants with specific, well-defined phenotypes.
Keywords: Clinical diagnostics; HPO; rare disease; variant database.
Figures

References
-
- Conter, C. , Rolland M. O., Cheillan D., Bonnet V., Maire I., and Froissart R.. 2006. Genetic heterogeneity of the GLDC gene in 28 unrelated patients with glycine encephalopathy. J. Inherit. Metab. Dis. 29:135–142. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
