

Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast
- PMID: 28117401
- PMCID: PMC5286201
- DOI: 10.1038/ncomms14061
Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast
Abstract
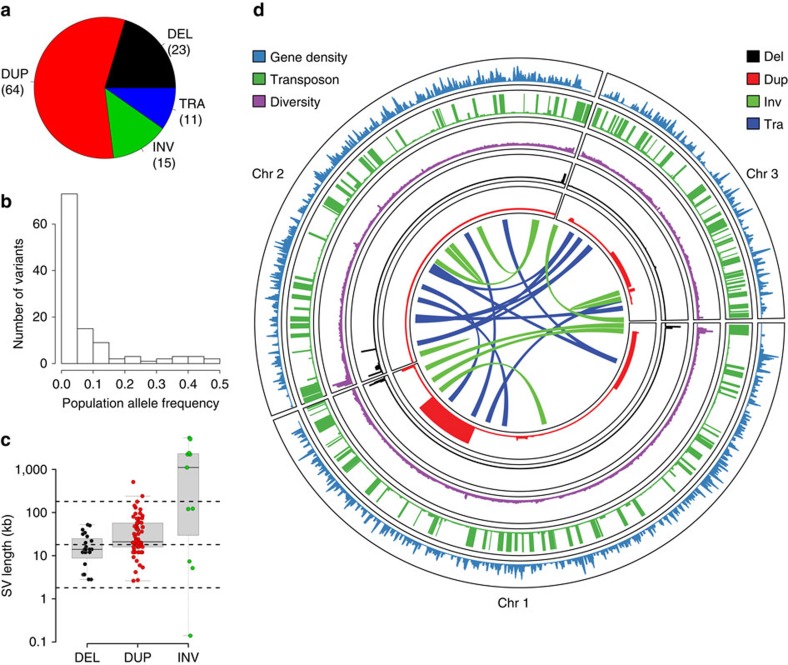
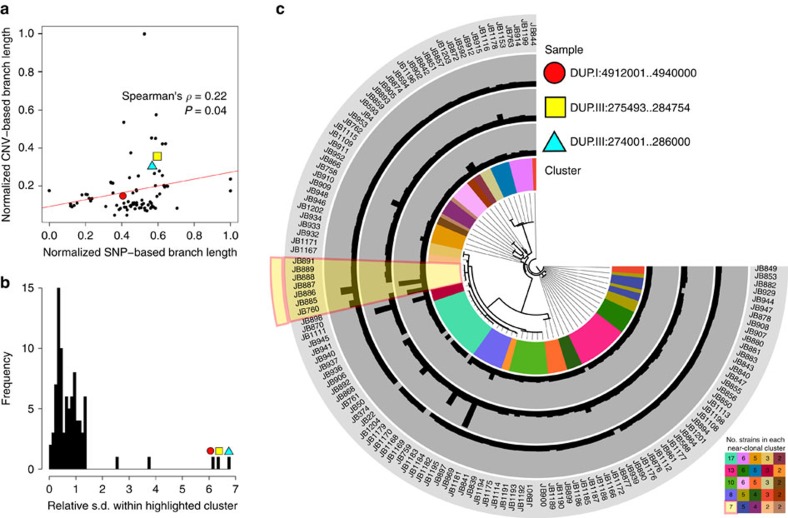
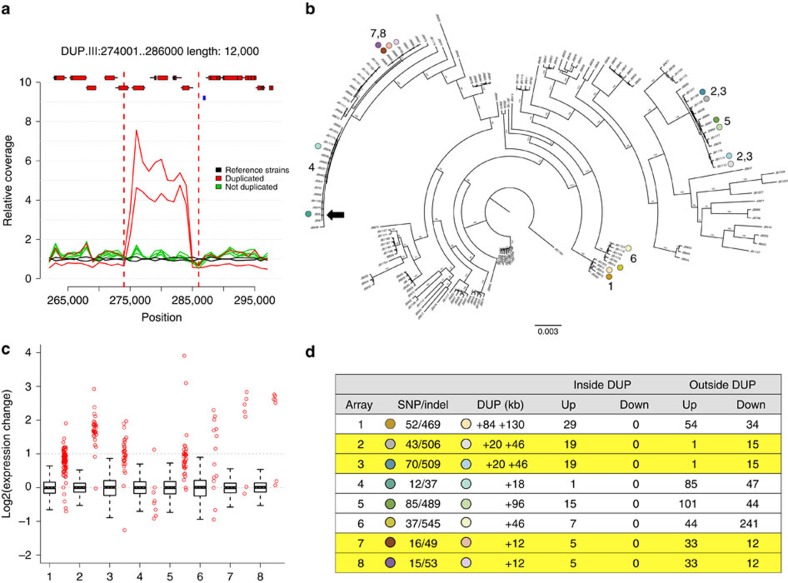
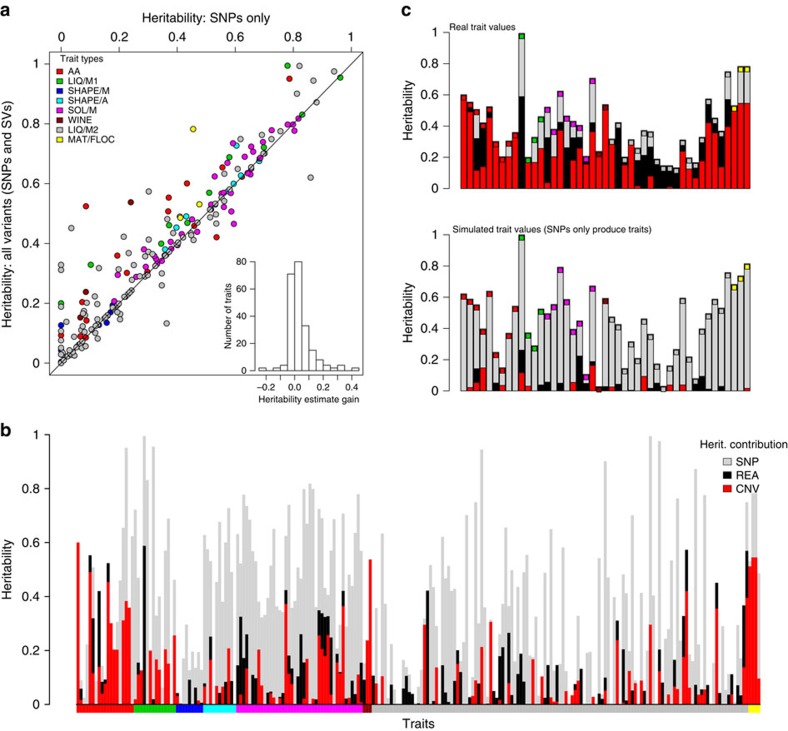
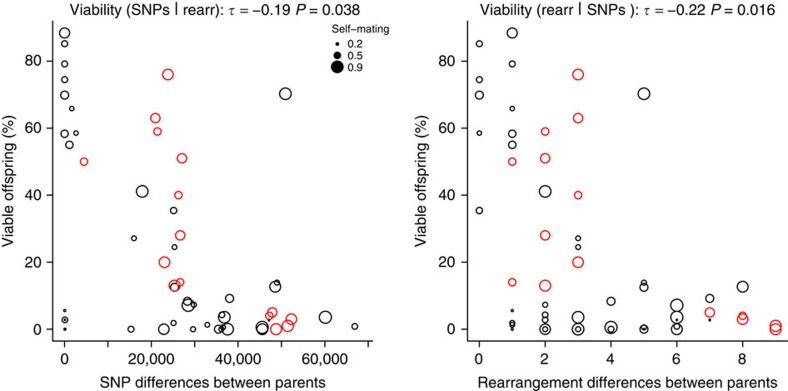
Large structural variations (SVs) within genomes are more challenging to identify than smaller genetic variants but may substantially contribute to phenotypic diversity and evolution. We analyse the effects of SVs on gene expression, quantitative traits and intrinsic reproductive isolation in the yeast Schizosaccharomyces pombe. We establish a high-quality curated catalogue of SVs in the genomes of a worldwide library of S. pombe strains, including duplications, deletions, inversions and translocations. We show that copy number variants (CNVs) show a variety of genetic signals consistent with rapid turnover. These transient CNVs produce stoichiometric effects on gene expression both within and outside the duplicated regions. CNVs make substantial contributions to quantitative traits, most notably intracellular amino acid concentrations, growth under stress and sugar utilization in winemaking, whereas rearrangements are strongly associated with reproductive isolation. Collectively, these findings have broad implications for evolution and for our understanding of quantitative traits including complex human diseases.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
