

Plasmodium malariae and P. ovale genomes provide insights into malaria parasite evolution
- PMID: 28117441
- PMCID: PMC5326575
- DOI: 10.1038/nature21038
Plasmodium malariae and P. ovale genomes provide insights into malaria parasite evolution
Abstract
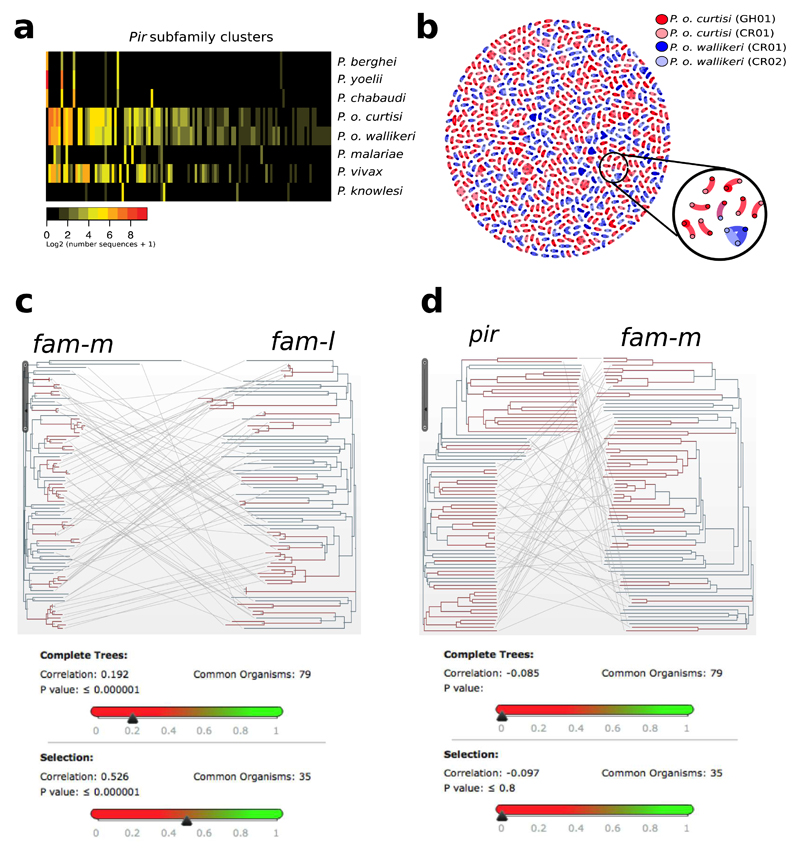
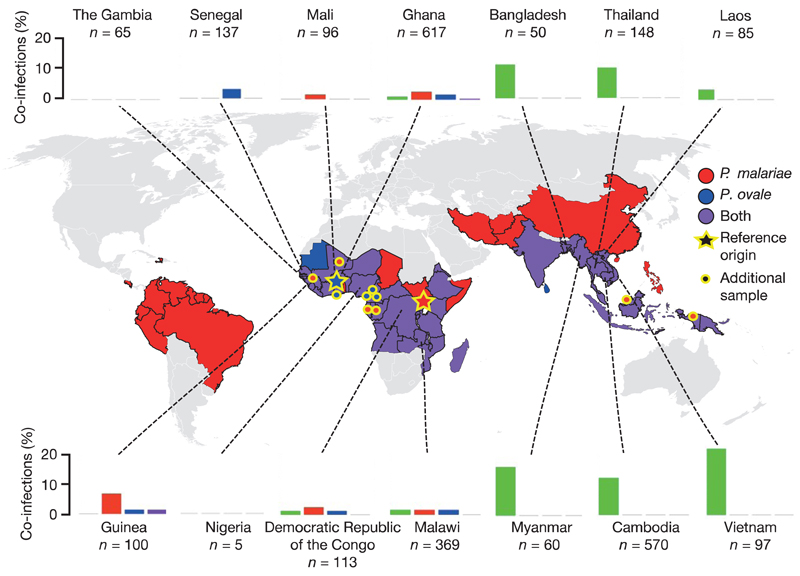
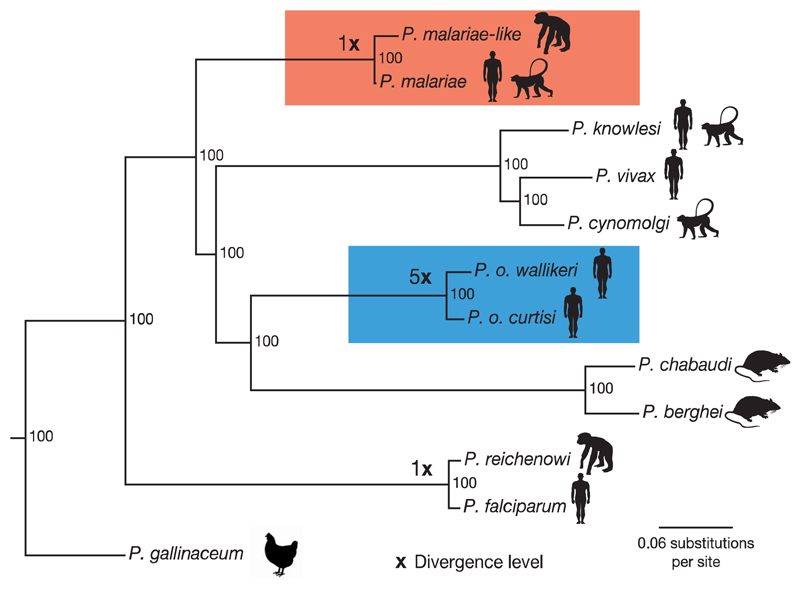
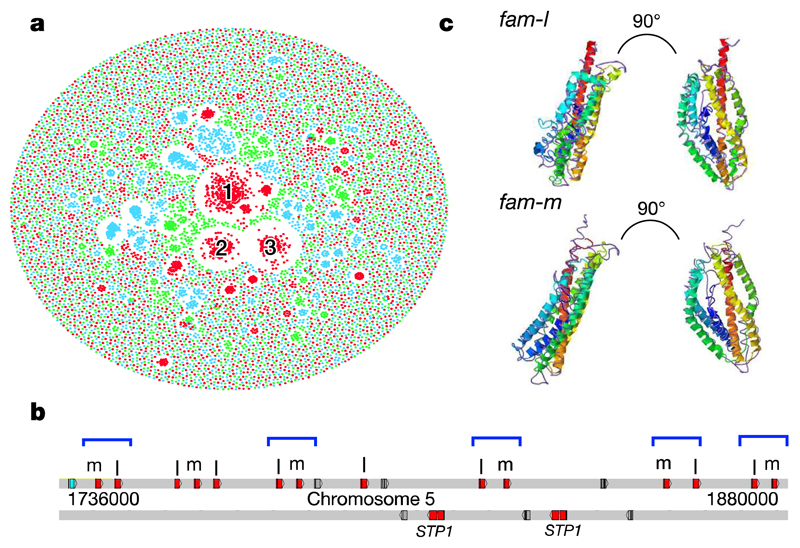
Elucidation of the evolutionary history and interrelatedness of Plasmodium species that infect humans has been hampered by a lack of genetic information for three human-infective species: P. malariae and two P. ovale species (P. o. curtisi and P. o. wallikeri). These species are prevalent across most regions in which malaria is endemic and are often undetectable by light microscopy, rendering their study in human populations difficult. The exact evolutionary relationship of these species to the other human-infective species has been contested. Using a new reference genome for P. malariae and a manually curated draft P. o. curtisi genome, we are now able to accurately place these species within the Plasmodium phylogeny. Sequencing of a P. malariae relative that infects chimpanzees reveals similar signatures of selection in the P. malariae lineage to another Plasmodium lineage shown to be capable of colonization of both human and chimpanzee hosts. Molecular dating suggests that these host adaptations occurred over similar evolutionary timescales. In addition to the core genome that is conserved between species, differences in gene content can be linked to their specific biology. The genome suggests that P. malariae expresses a family of heterodimeric proteins on its surface that have structural similarities to a protein crucial for invasion of red blood cells. The data presented here provide insight into the evolution of the Plasmodium genus as a whole.
Figures
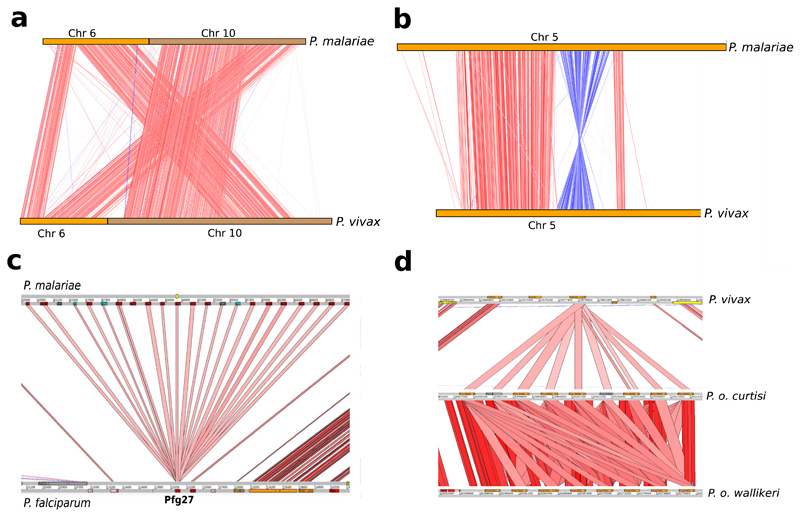
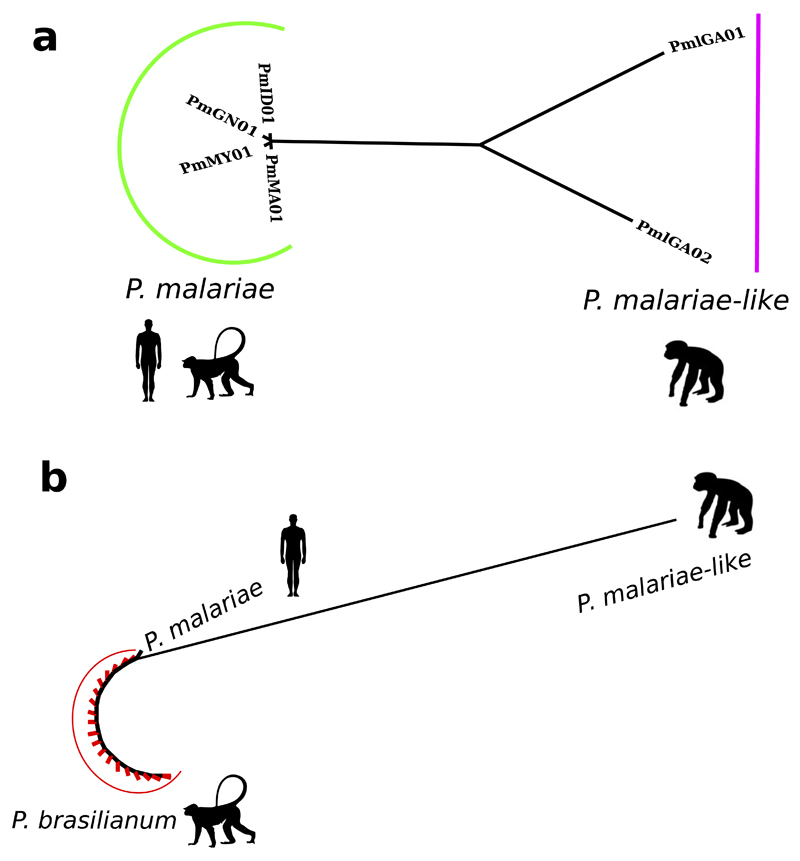
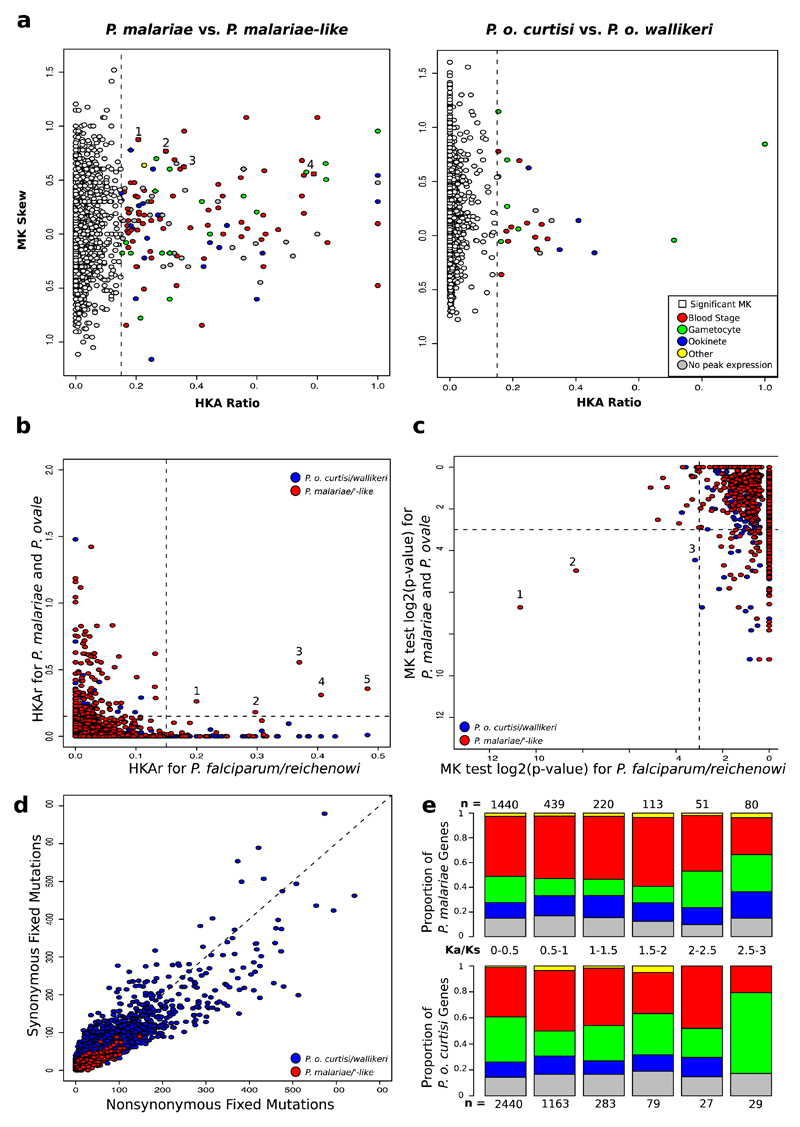
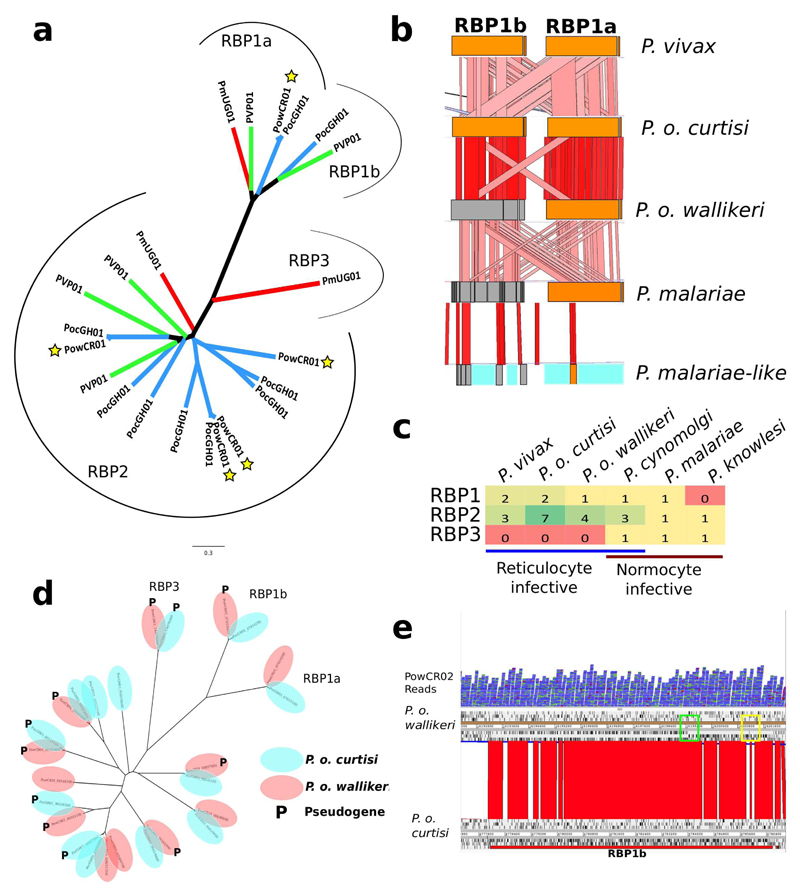
Comment in
-
A Feast of Malaria Parasite Genomes.Cell Host Microbe. 2017 Mar 8;21(3):310-312. doi: 10.1016/j.chom.2017.02.019. Cell Host Microbe. 2017. PMID: 28279339 Free PMC article.
References
-
- Sutherland CJ, et al. Two nonrecombining sympatric forms of the human malaria parasite Plasmodium ovale occur globally. J Infect Dis. 2010;201:1544–1550. - PubMed
-
- Bousema T, Okell L, Felger I, Drakeley C. Asymptomatic malaria infections: detectability, transmissibility and public health relevance. Nat Rev Microbiol. 2014;12:833–840. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
