

Aortopathy in a Mouse Model of Marfan Syndrome Is Not Mediated by Altered Transforming Growth Factor β Signaling
- PMID: 28119285
- PMCID: PMC5523644
- DOI: 10.1161/JAHA.116.004968
Aortopathy in a Mouse Model of Marfan Syndrome Is Not Mediated by Altered Transforming Growth Factor β Signaling
Abstract
Background: Marfan syndrome (MFS) is caused by mutations in the gene encoding fibrillin-1 (FBN1); however, the mechanisms through which fibrillin-1 deficiency causes MFS-associated aortopathy are uncertain. Recently, attention was focused on the hypothesis that MFS-associated aortopathy is caused by increased transforming growth factor-β (TGF-β) signaling in aortic medial smooth muscle cells (SMC). However, there are many reasons to doubt that TGF-β signaling drives MFS-associated aortopathy. We used a mouse model to test whether SMC TGF-β signaling is perturbed by a fibrillin-1 variant that causes MFS and whether blockade of SMC TGF-β signaling prevents MFS-associated aortopathy.
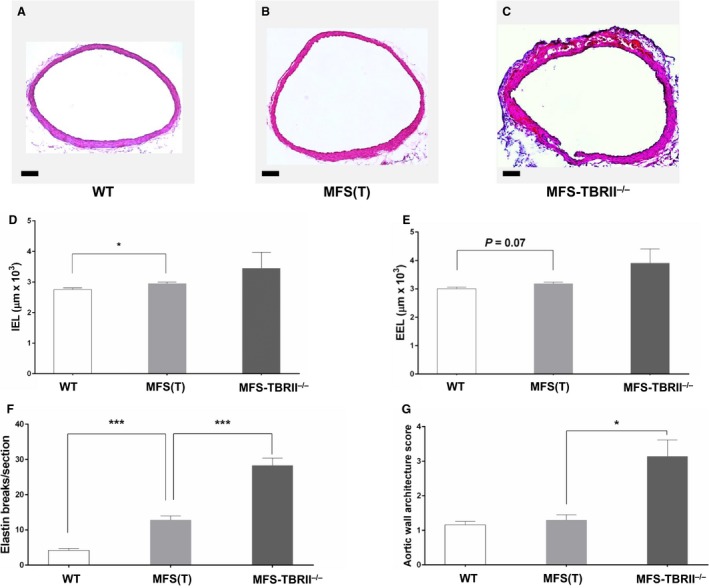
Methods and results: MFS mice (Fbn1C1039G/+ genotype) were genetically modified to allow postnatal SMC-specific deletion of the type II TGF-β receptor (TBRII; essential for physiologic TGF-β signaling). In young MFS mice with and without superimposed deletion of SMC-TBRII, we measured aortic dimensions, histopathology, activation of aortic SMC TGF-β signaling pathways, and changes in aortic SMC gene expression. Young Fbn1C1039G/+ mice had ascending aortic dilation and significant disruption of aortic medial architecture. Both aortic dilation and disrupted medial architecture were exacerbated by superimposed deletion of TBRII. TGF-β signaling was unaltered in aortic SMC of young MFS mice; however, SMC-specific deletion of TBRII in Fbn1C1039G/+ mice significantly decreased activation of SMC TGF-β signaling pathways.
Conclusions: In young Fbn1C1039G/+ mice, aortopathy develops in the absence of detectable alterations in SMC TGF-β signaling. Loss of physiologic SMC TGF-β signaling exacerbates MFS-associated aortopathy. Our data support a protective role for SMC TGF-β signaling during early development of MFS-associated aortopathy.
Keywords: Marfan syndrome; fibrillin‐1; gene expression; genetically altered mice; signaling pathways; transforming growth factor‐β pathway aneurysm.
© 2017 The Authors. Published on behalf of the American Heart Association, Inc., by Wiley Blackwell.
Figures






Comment in
-
Transforming Growth Factor-β in Thoracic Aortic Aneurysms: Good, Bad, or Irrelevant?J Am Heart Assoc. 2017 Jan 24;6(1):e005221. doi: 10.1161/JAHA.116.005221. J Am Heart Assoc. 2017. PMID: 28119286 Free PMC article. No abstract available.
References
-
- Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, Stetten G, Meyers DA, Francomano CA. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. - PubMed
-
- Pyeritz RE. The Marfan syndrome. Annu Rev Med. 2000;51:481–510. - PubMed
-
- McKusick VA. The defect in Marfan syndrome. Nature. 1991;352:279–281. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
