

EZH2 or HDAC1 Inhibition Reverses Multiple Myeloma-Induced Epigenetic Suppression of Osteoblast Differentiation
- PMID: 28119431
- PMCID: PMC5524543
- DOI: 10.1158/1541-7786.MCR-16-0242-T
EZH2 or HDAC1 Inhibition Reverses Multiple Myeloma-Induced Epigenetic Suppression of Osteoblast Differentiation
Abstract
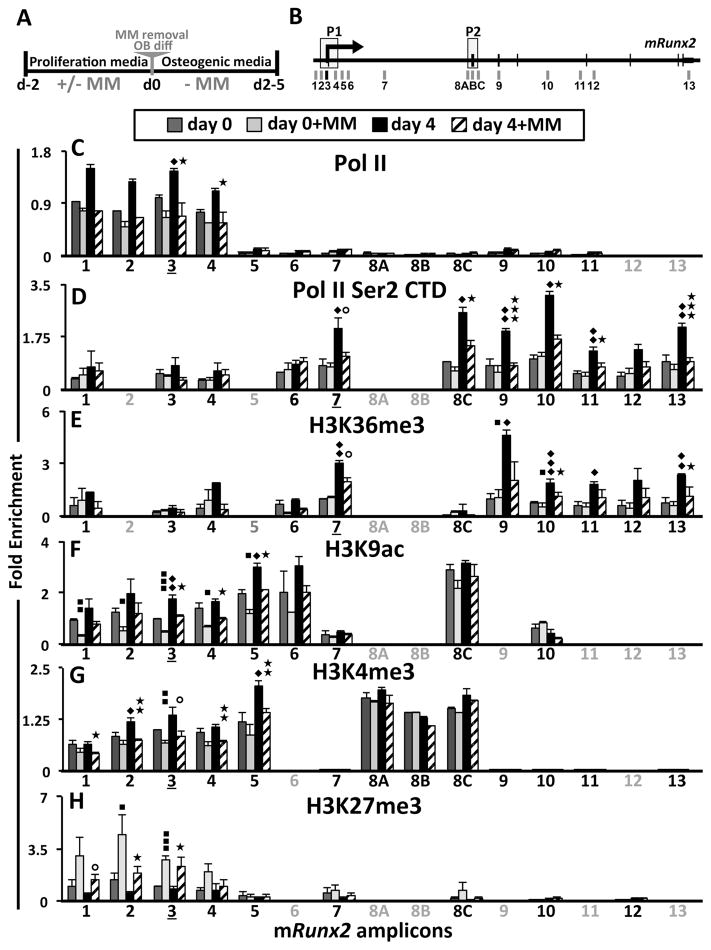
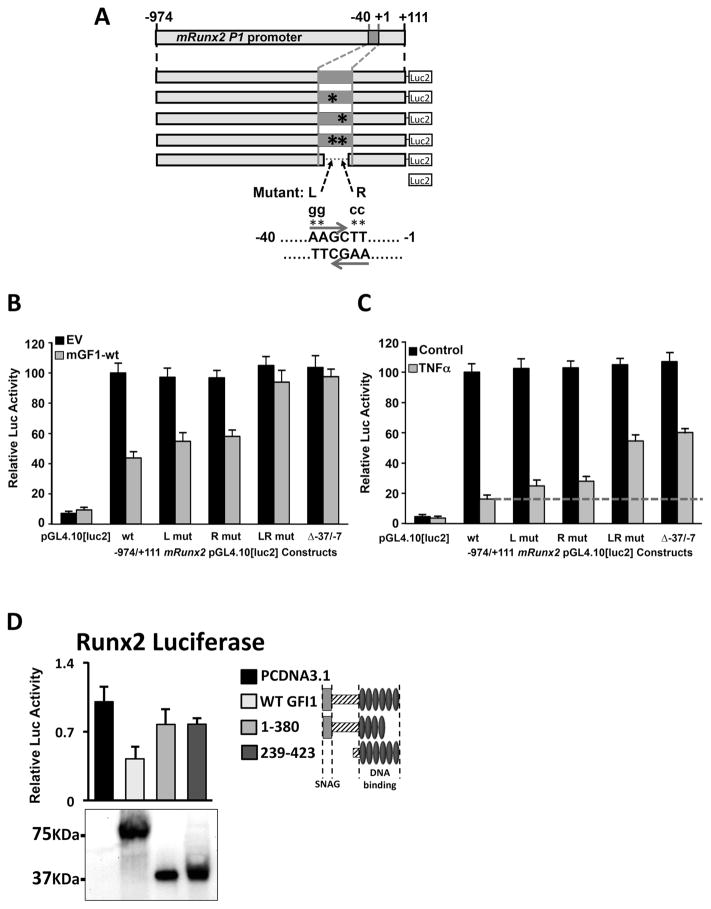
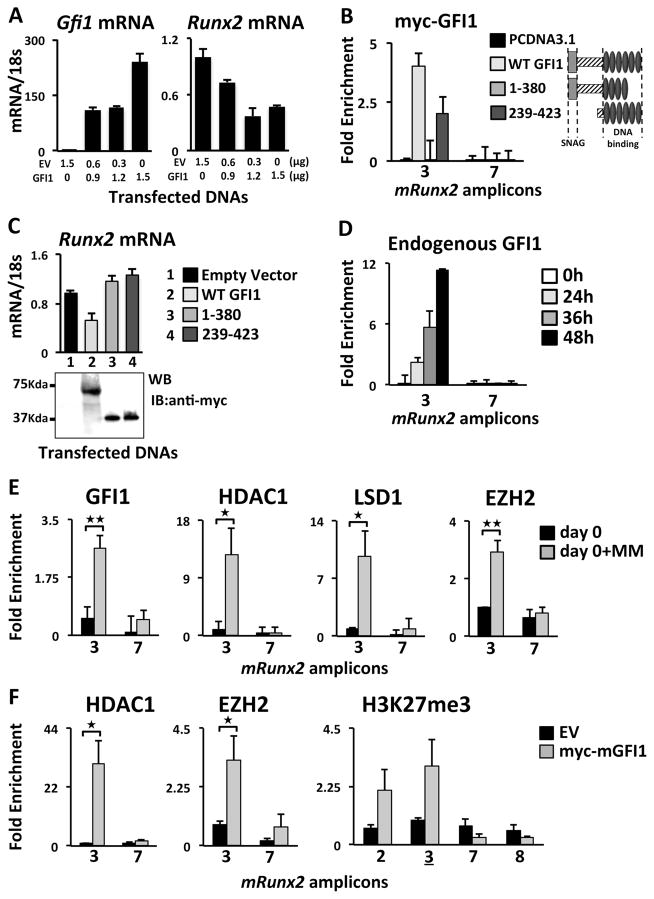
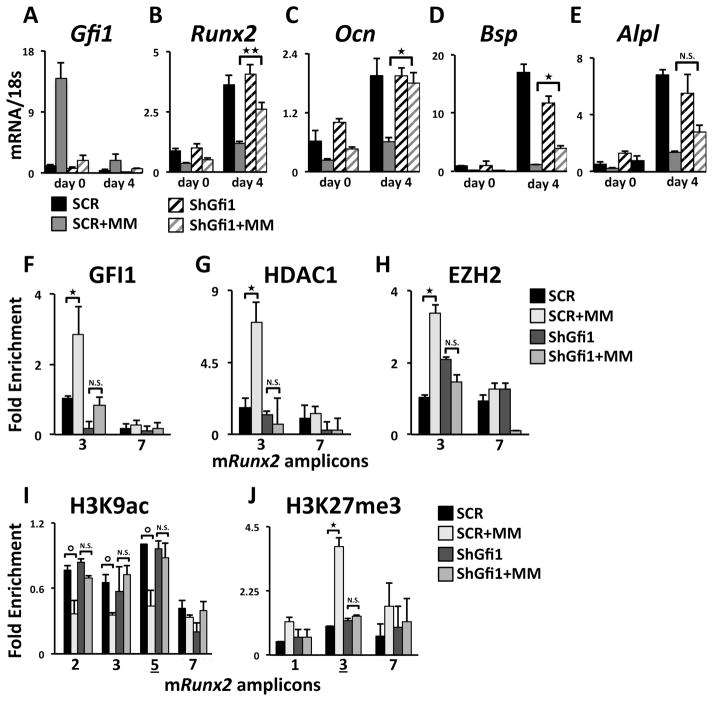
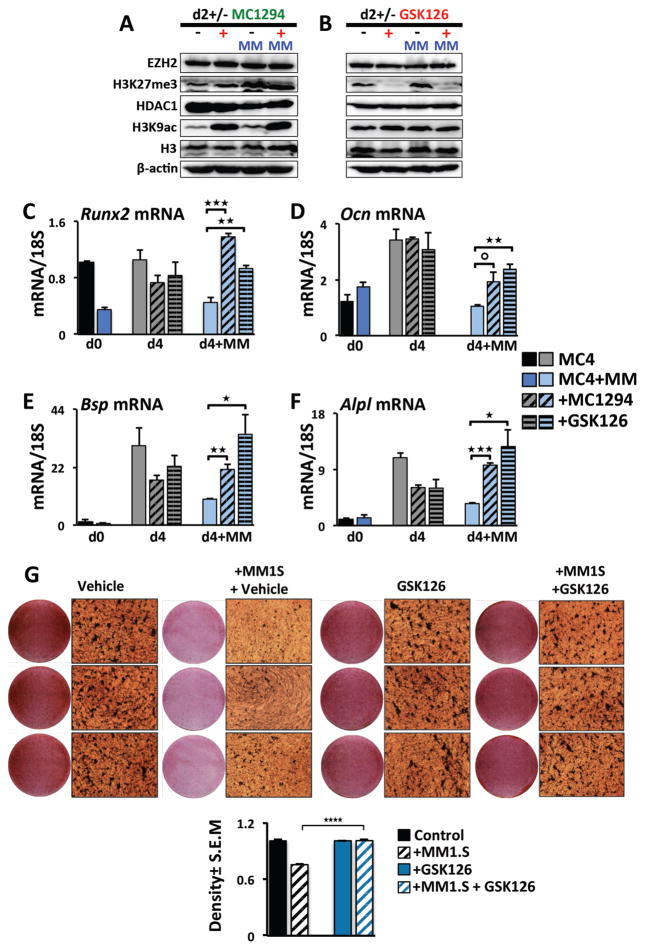
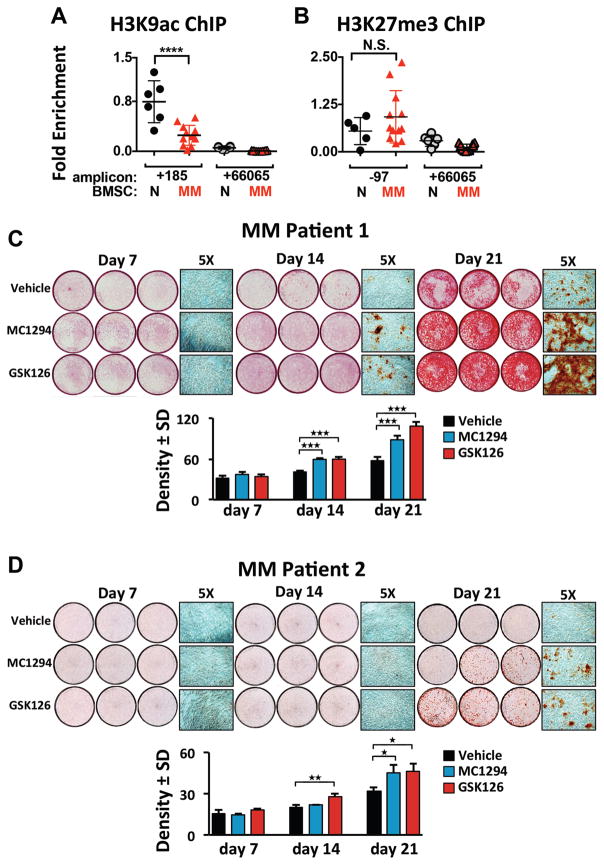
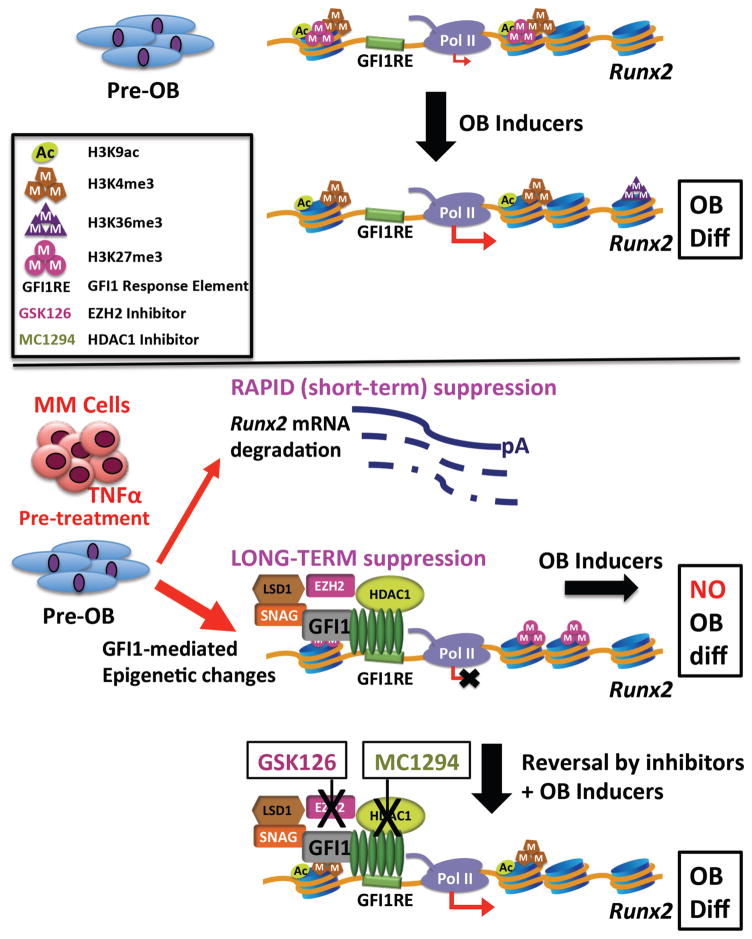
In multiple myeloma, osteolytic lesions rarely heal because of persistent suppressed osteoblast differentiation resulting in a high fracture risk. Herein, chromatin immunoprecipitation analyses reveal that multiple myeloma cells induce repressive epigenetic histone changes at the Runx2 locus that prevent osteoblast differentiation. The most pronounced multiple myeloma-induced changes were at the Runx2-P1 promoter, converting it from a poised bivalent state to a repressed state. Previously, it was observed that multiple myeloma induces the transcription repressor GFI1 in osteoblast precursors, which correlates with decreased Runx2 expression, thus prompting detailed characterization of the multiple myeloma and TNFα-dependent GFI1 response element within the Runx2-P1 promoter. Further analyses reveal that multiple myeloma-induced GFI1 binding to Runx2 in osteoblast precursors and recruitment of the histone modifiers HDAC1, LSD1, and EZH2 is required to establish and maintain Runx2 repression in osteogenic conditions. These GFI1-mediated repressive chromatin changes persist even after removal of multiple myeloma. Ectopic GFI1 is sufficient to bind to Runx2, recruit HDAC1 and EZH2, increase H3K27me3 on the gene, and prevent osteogenic induction of endogenous Runx2 expression. Gfi1 knockdown in MC4 cells blocked multiple myeloma-induced recruitment of HDAC1 and EZH2 to Runx2, acquisition of repressive chromatin architecture, and suppression of osteoblast differentiation. Importantly, inhibition of EZH2 or HDAC1 activity in pre-osteoblasts after multiple myeloma exposure in vitro or in osteoblast precursors from patients with multiple myeloma reversed the repressive chromatin architecture at Runx2 and rescued osteoblast differentiation.Implications: This study suggests that therapeutically targeting EZH2 or HDAC1 activity may reverse the profound multiple myeloma-induced osteoblast suppression and allow repair of the lytic lesions. Mol Cancer Res; 15(4); 405-17. ©2017 AACR.
©2017 American Association for Cancer Research.
Conflict of interest statement
Figures
References
-
- Roodman GD. Pathogenesis of myeloma bone disease. J Cell Biochem. 2010;109:283–91. - PubMed
-
- Saad F, Lipton A, Cook R, Chen YM, Smith M, Coleman R. Pathologic fractures correlate with reduced survival in patients with malignant bone disease. Cancer. 2007;110:1860–7. - PubMed
-
- Giuliani N, Rizzoli V, Roodman GD. Multiple myeloma bone disease: Pathophysiology of osteoblast inhibition. Blood. 2006;108:3992–6. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
