

Dual Role of Epidermal Growth Factor Receptor in Liver Injury and Regeneration after Acetaminophen Overdose in Mice
- PMID: 28123000
- PMCID: PMC6020708
- DOI: 10.1093/toxsci/kfw213
Dual Role of Epidermal Growth Factor Receptor in Liver Injury and Regeneration after Acetaminophen Overdose in Mice
Abstract
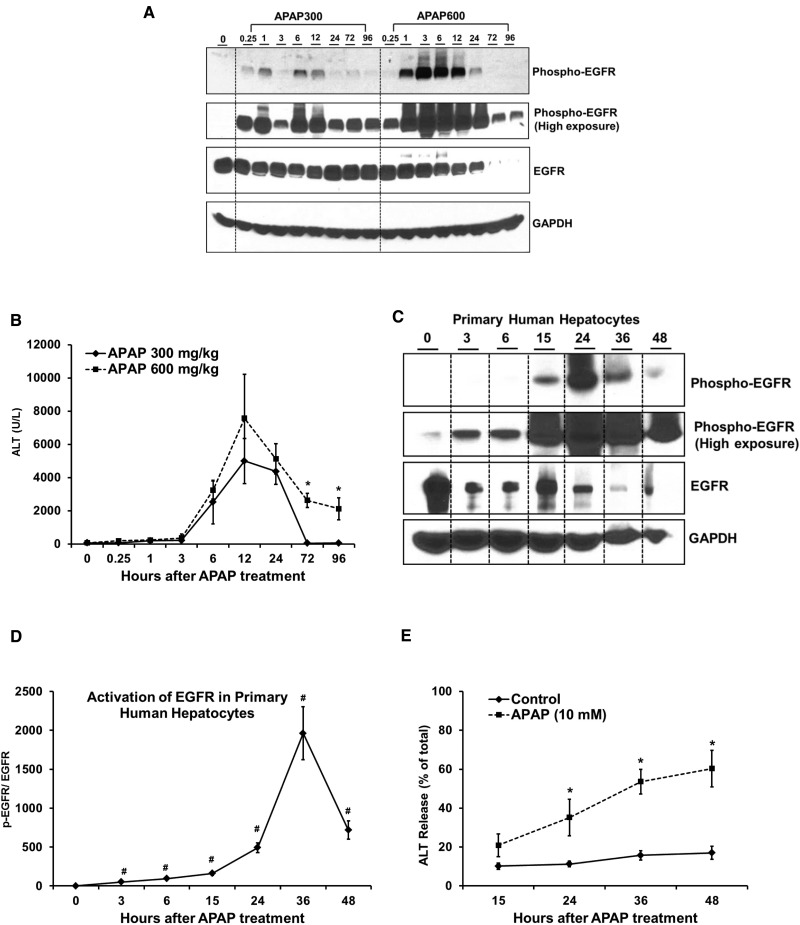
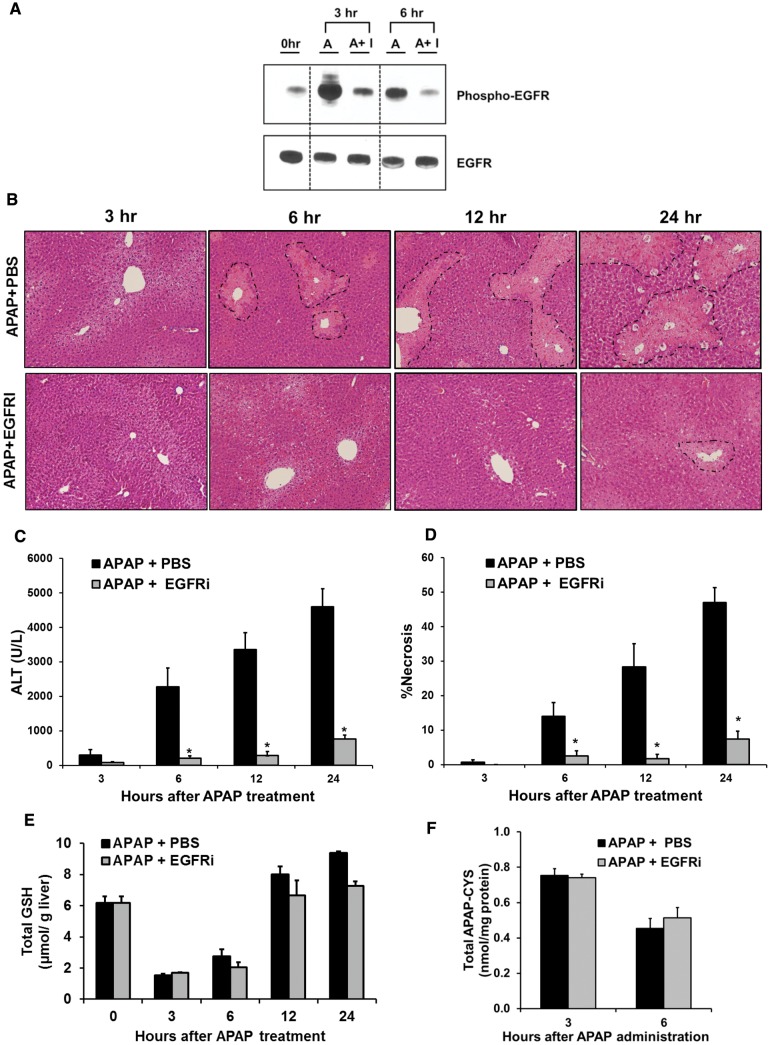
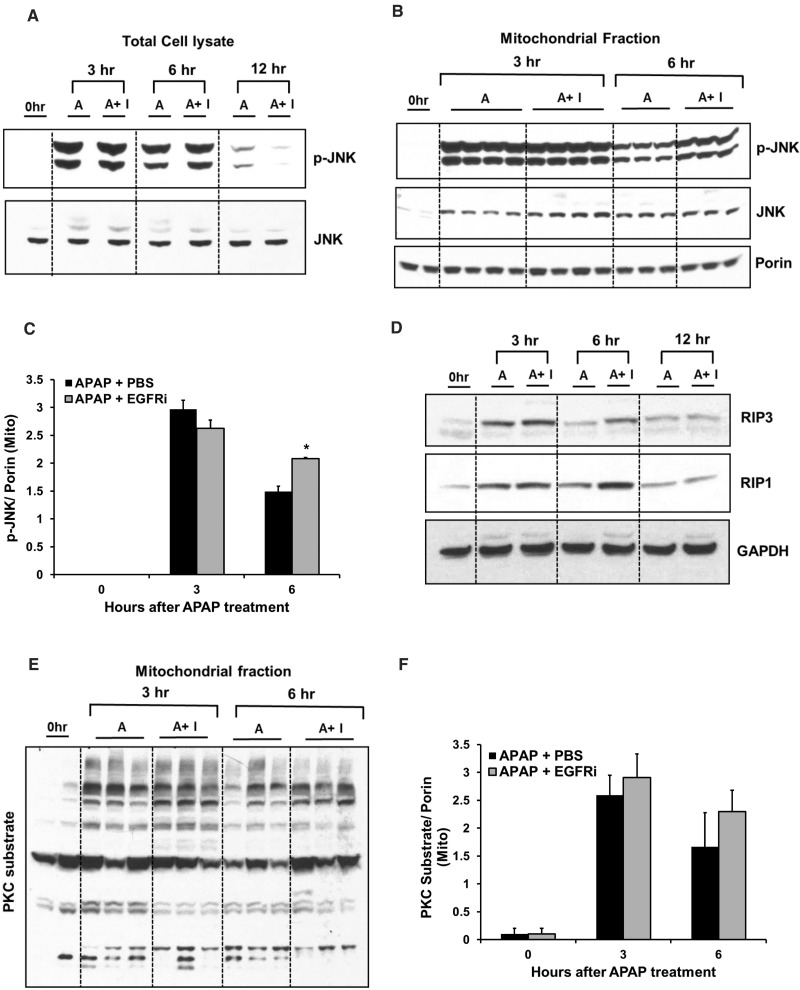
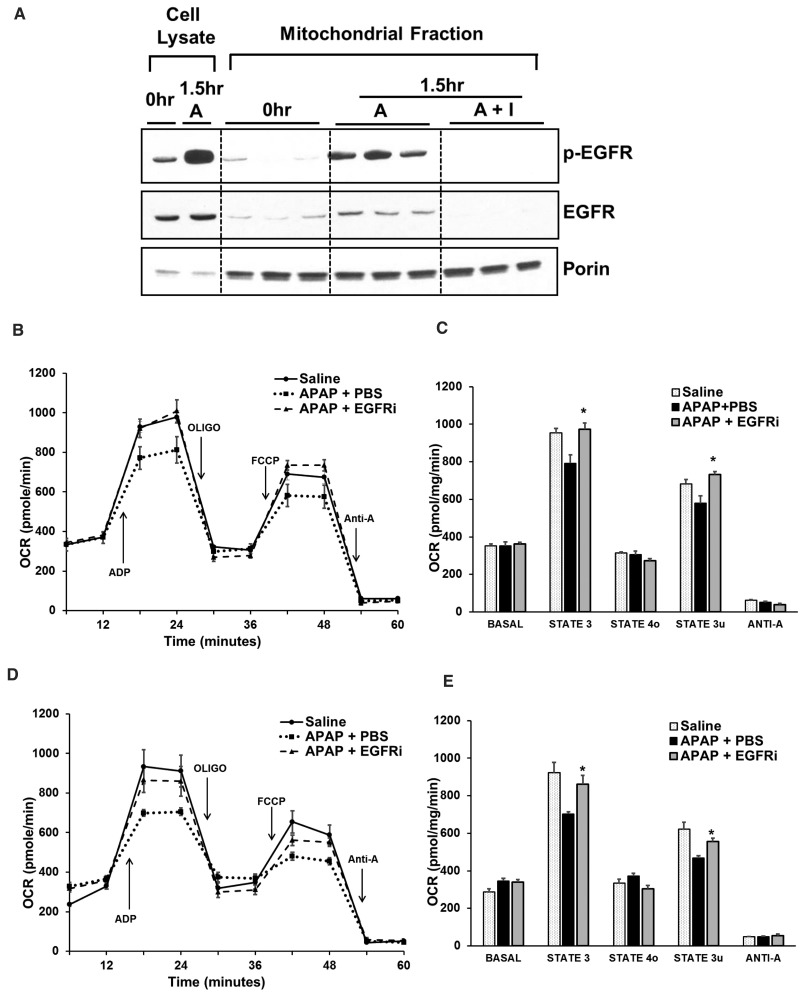
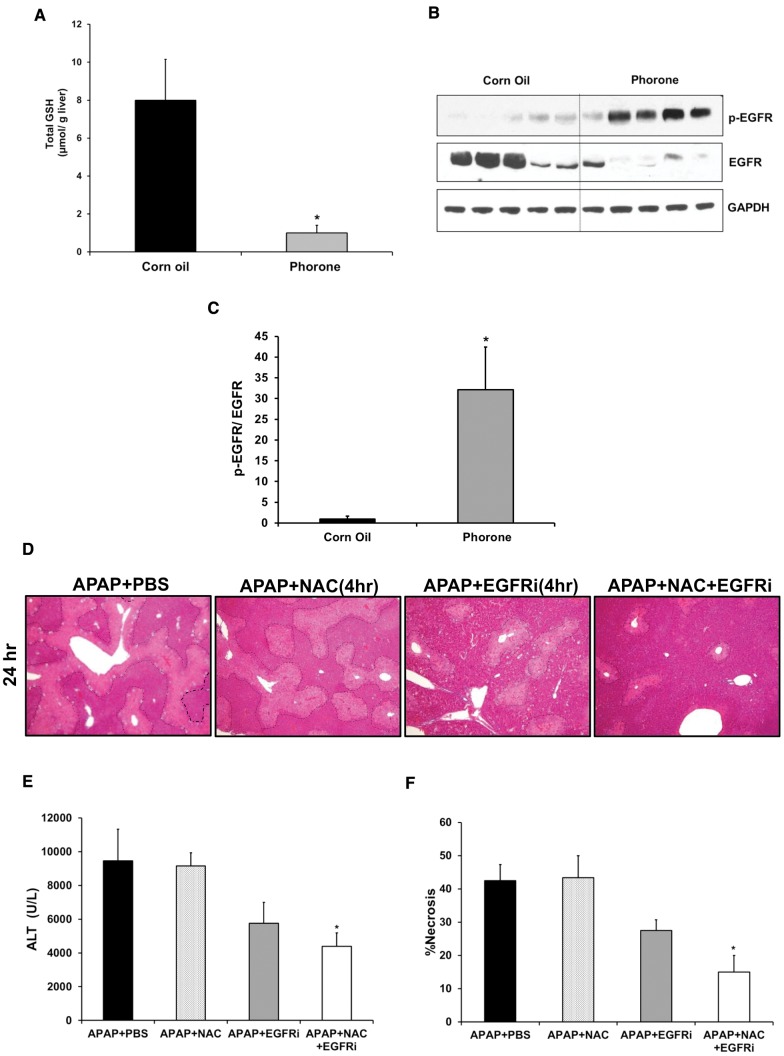
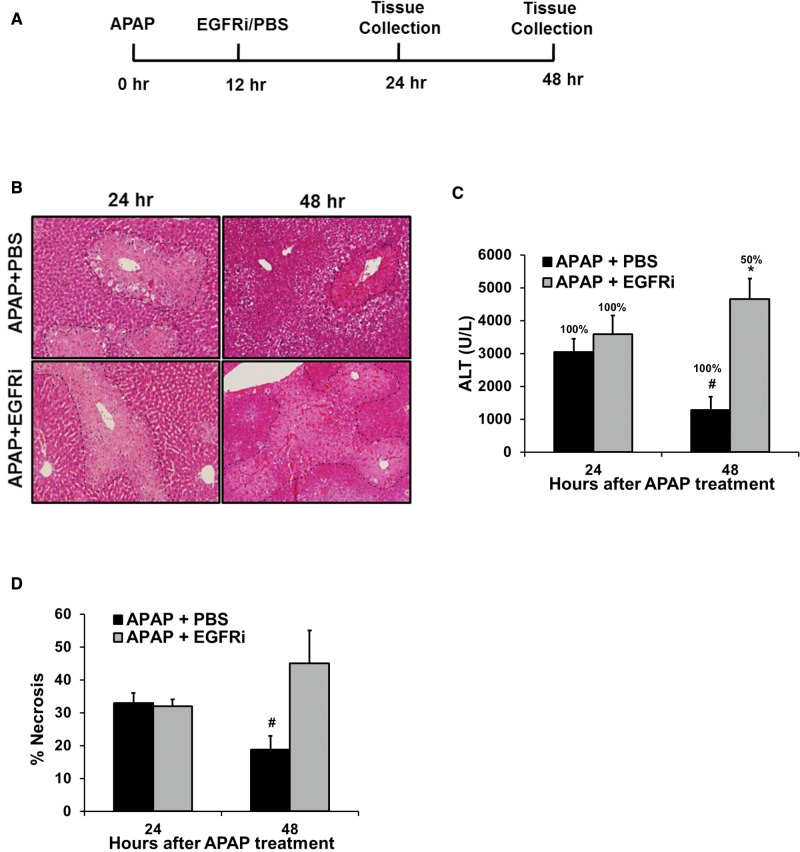
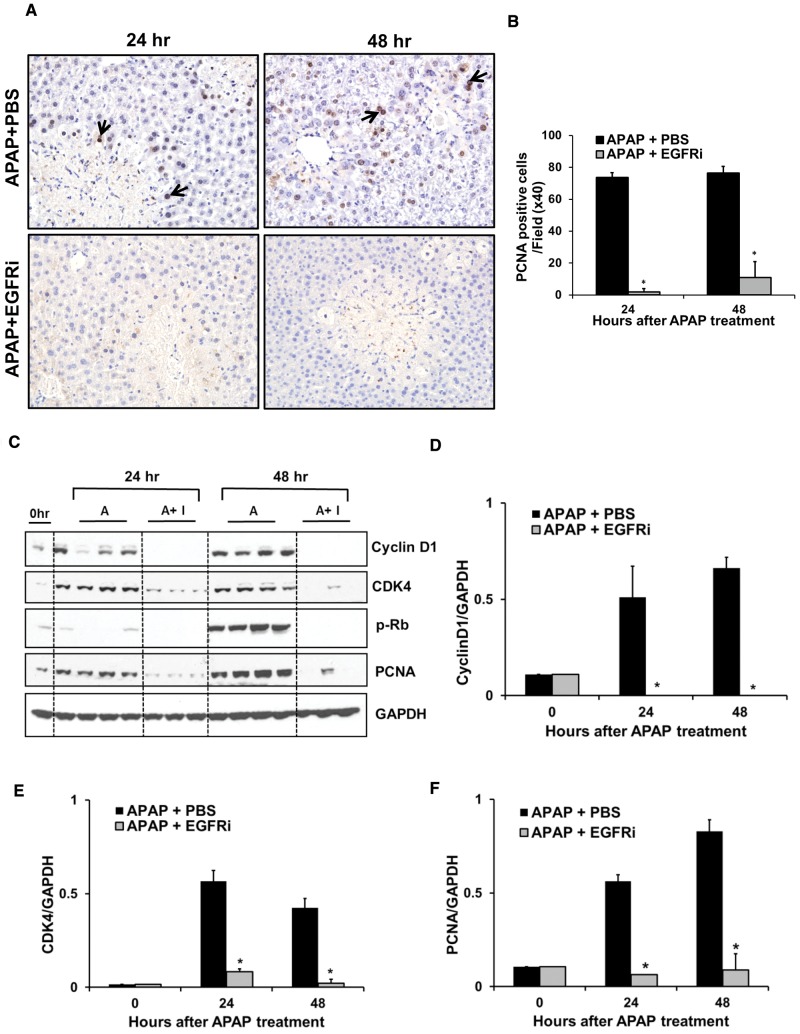
Epidermal growth factor receptor (EGFR) plays a crucial role in hepatocyte proliferation. Its role in acetaminophen (APAP)-mediated hepatotoxicity and subsequent liver regeneration is completely unknown. Role of EGFR after APAP-overdose in mice was studied using pharmacological inhibition strategy. Rapid, sustained and dose-dependent activation of EGFR was noted after APAP-treatment in mice, which was triggered by glutathione depletion. EGFR-activation was also observed in primary human hepatocytes after APAP-treatment, preceding elevation of toxicity markers. Treatment of mice with an EGFR-inhibitor (EGFRi), Canertinib, 1h post-APAP resulted in robust inhibition of EGFR-activation and a striking reduction in APAP-induced liver injury. Metabolic activation of APAP, formation of APAP-protein adducts, APAP-mediated JNK-activation and its mitochondrial translocation were not altered by EGFRi. Interestingly, EGFR rapidly translocated to mitochondria after APAP-treatment. EGFRi-treatment abolished mitochondrial EGFR activity, prevented APAP-mediated mitochondrial dysfunction/oxidative-stress and release of endonucleases from mitochondria, which are responsible for DNA-damage/necrosis. Treatment with N-acetylcysteine (NAC), 4h post-APAP in mice did not show any protection but treatment of EGFRi in combination with NAC showed decrease in liver injury. Finally, delayed treatment with EGFRi, 12-h post-APAP, did not alter peak injury but caused impairment of liver regeneration resulting in sustained injury and decreased survival after APAP overdose in mice. Impairment of regeneration was due to inhibition of cyclinD1 induction and cell cycle arrest. Our study has revealed a new dual role of EGFR both in initiation of APAP-injury and in stimulation of subsequent compensatory regeneration after APAP-overdose.
Keywords: Cyclin D1; Hepatotoxicity; N-acetylcysteine; hepatocyte proliferation.; mitochondrial dysfunction.
© The Author 2016. Published by Oxford University Press on behalf of the Society of Toxicology. All rights reserved. For Permissions, please e-mail: journals.permissions@oup.com.
Figures

References
-
- Arany I. (2008). Dual role of the activated epidermal growth factor receptor in renal tubular cells during stress. Kidney Int. 73, 5–7. - PubMed
-
- Arany I., Megyesi J. K., Kaneto H., Price P. M., Safirstein R. L. (2004). Cisplatin-induced cell death is EGFR/src/ERK signaling dependent in mouse proximal tubule cells. Am. J. Physiol. Renal Physiol. 287, F543–F549. - PubMed
-
- Bernal W., Lee W. M., Wendon J., Larsen F. S., Williams R. (2015). Acute liver failure: A curable disease by 2024?. J. Hepatol. 62(1 Suppl), S112–S120., - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
