

Tracing the dynamics of gene transcripts after organismal death
- PMID: 28123054
- PMCID: PMC5303275
- DOI: 10.1098/rsob.160267
Tracing the dynamics of gene transcripts after organismal death
Abstract
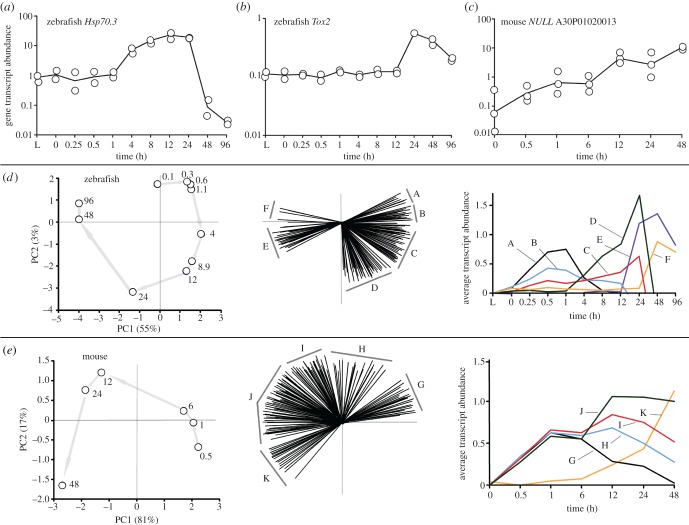
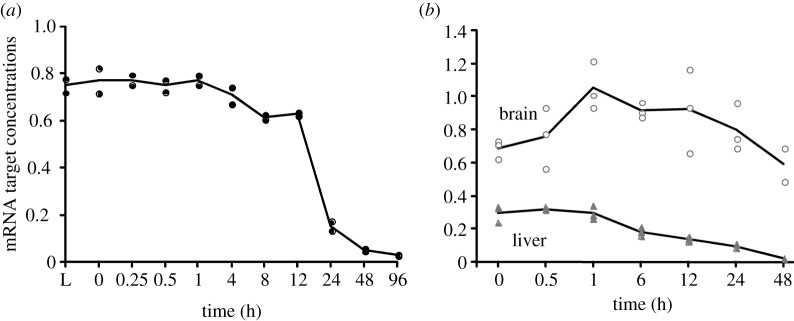
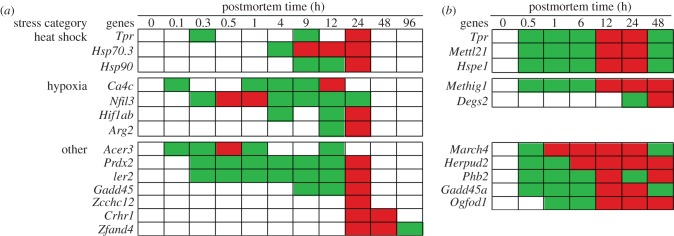
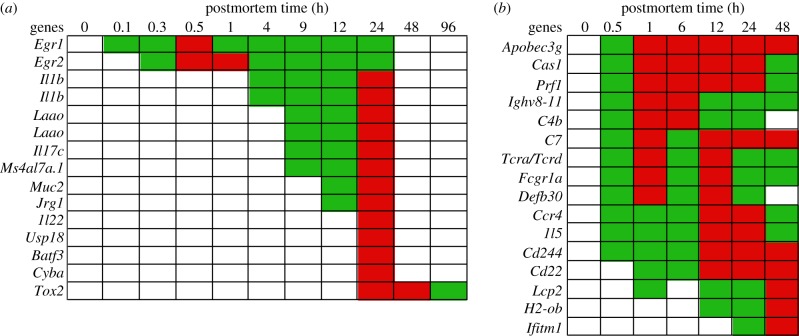
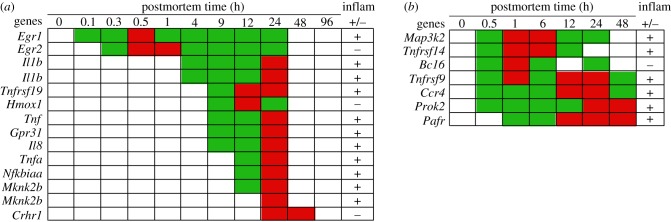
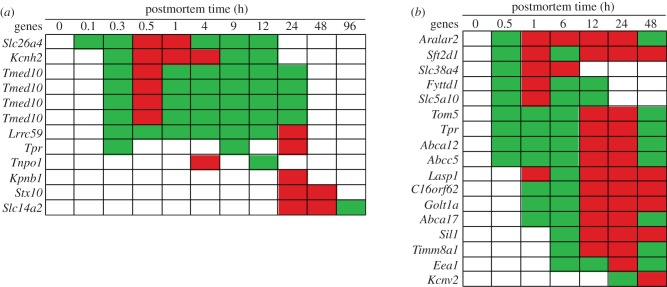
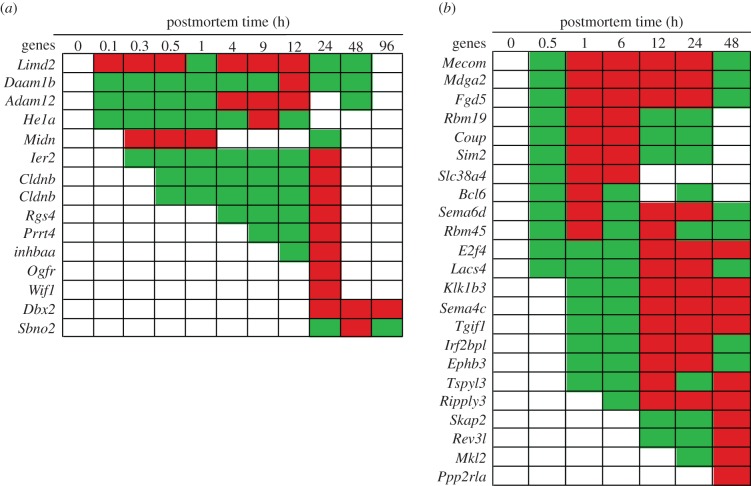
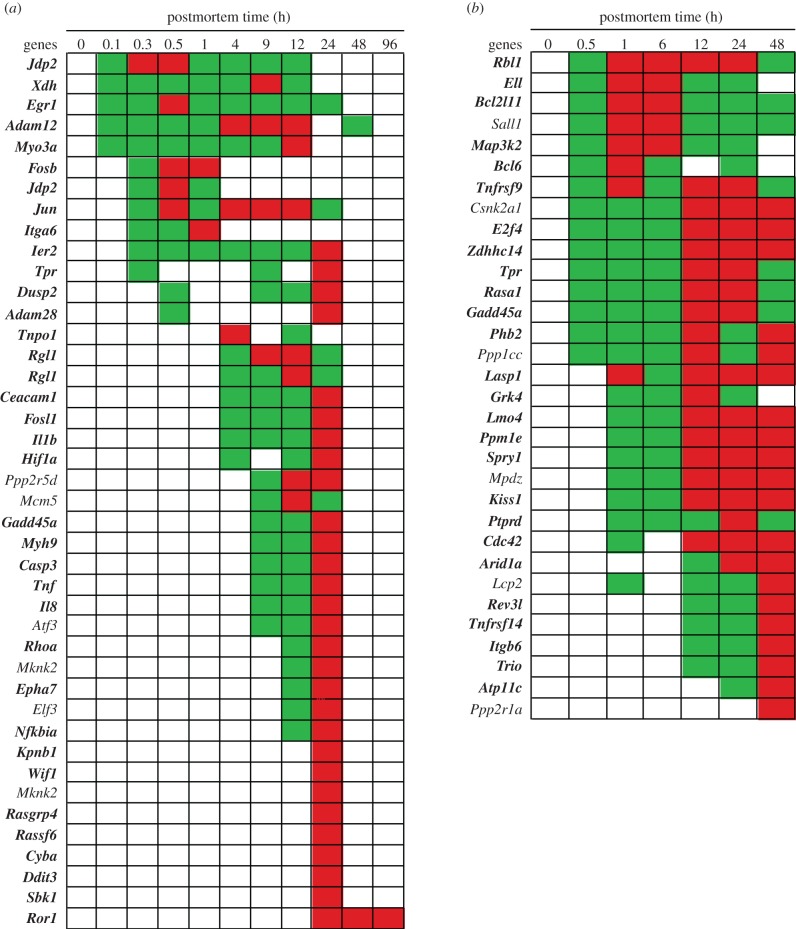
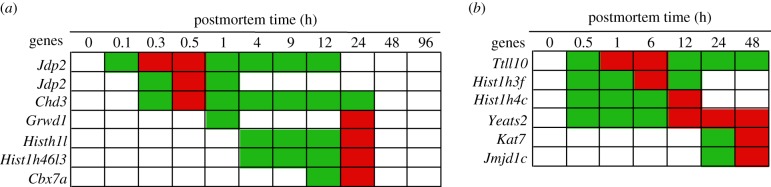
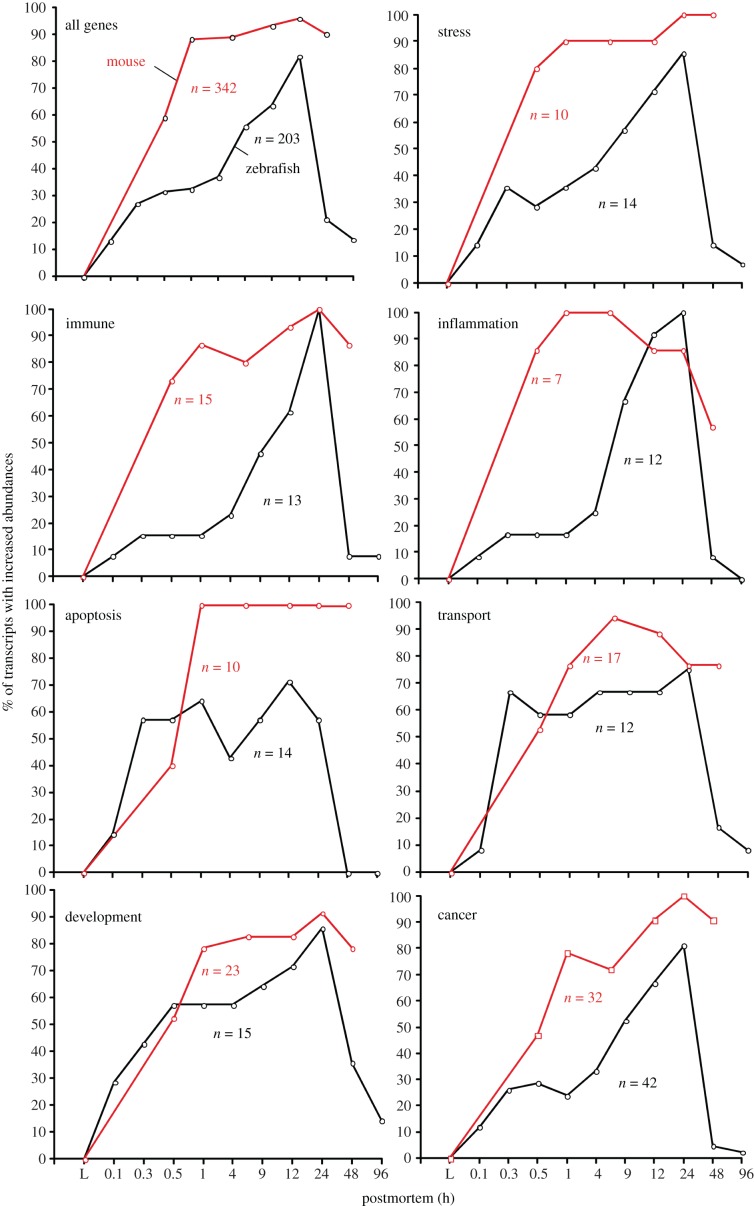
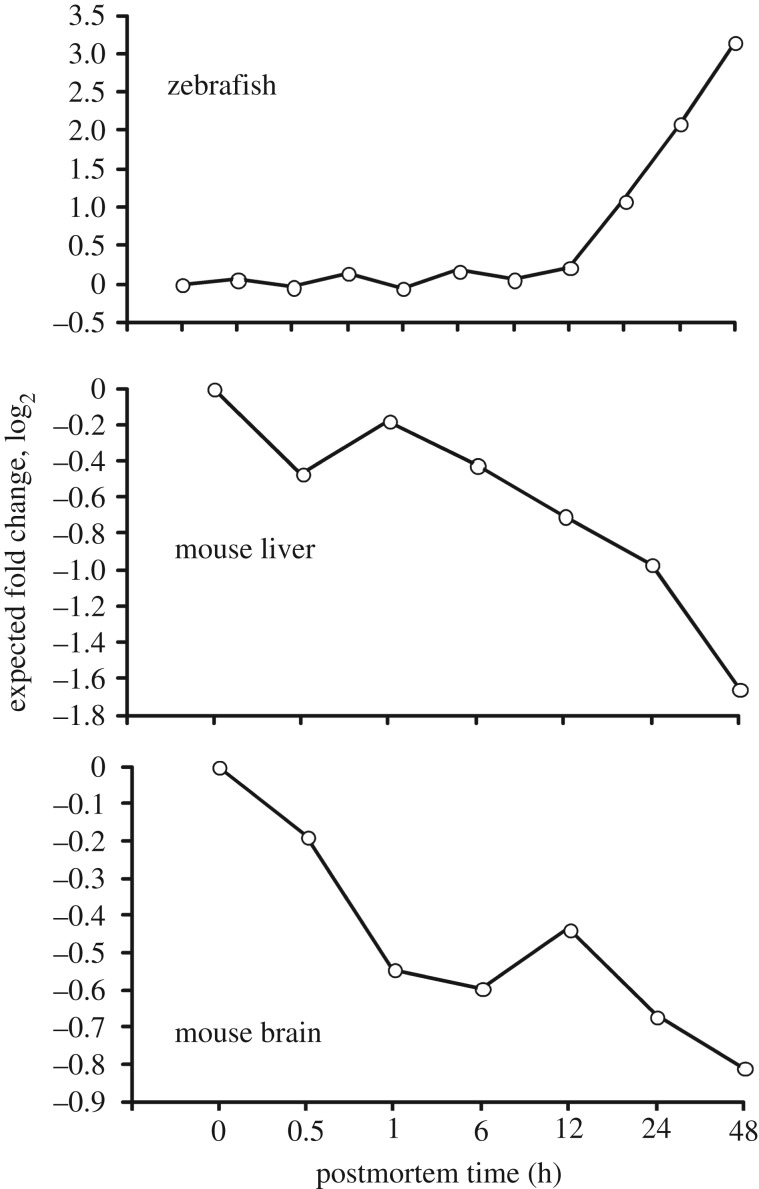
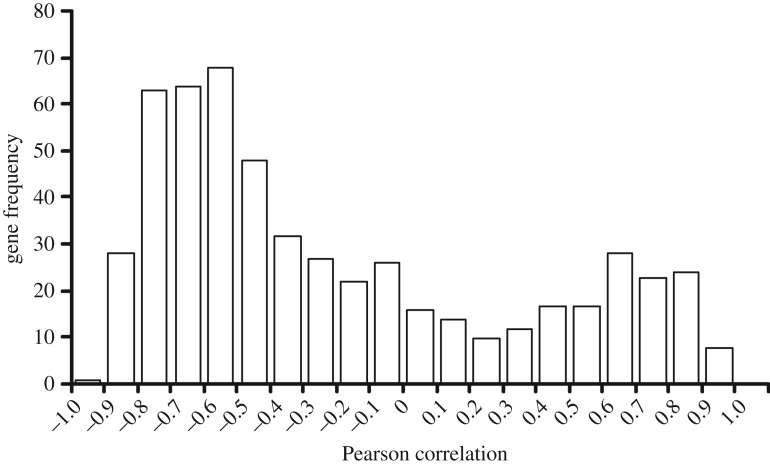
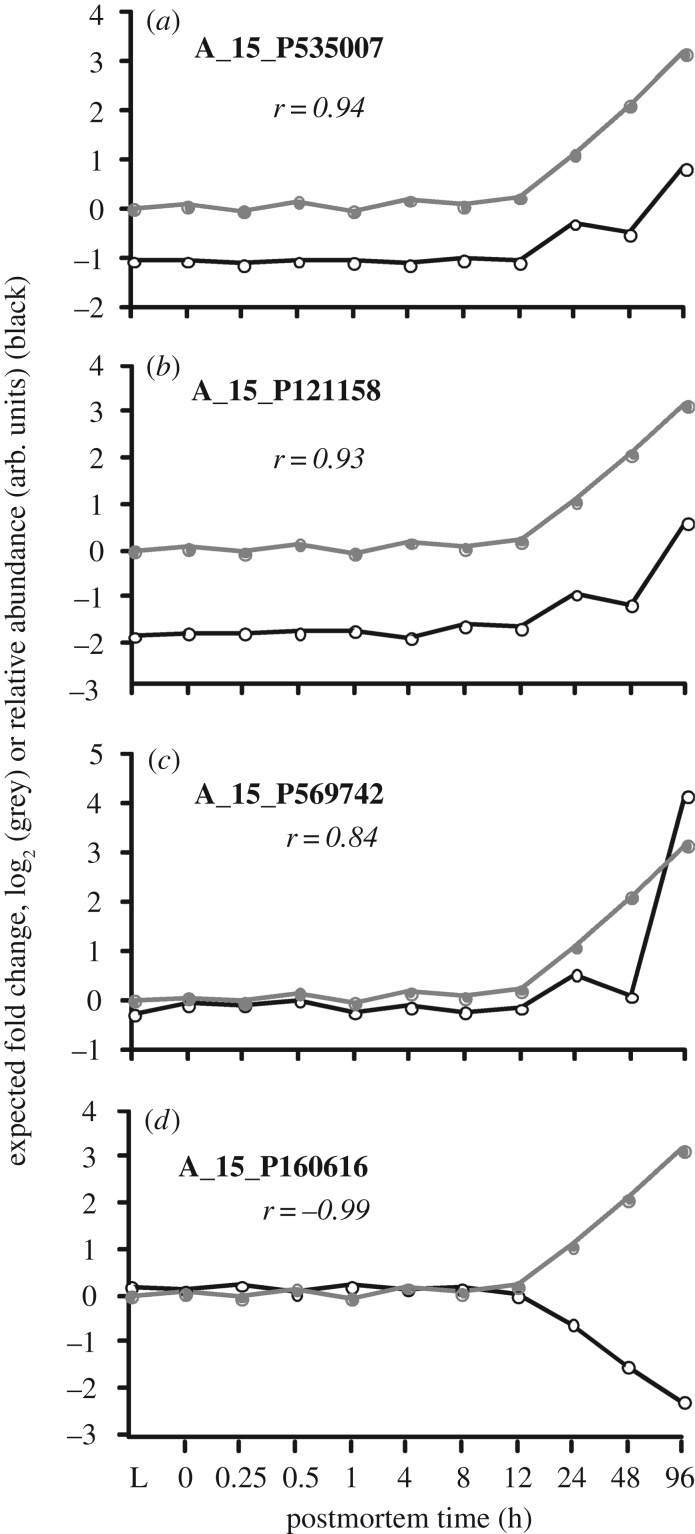
In life, genetic and epigenetic networks precisely coordinate the expression of genes-but in death, it is not known if gene expression diminishes gradually or abruptly stops or if specific genes and pathways are involved. We studied this by identifying mRNA transcripts that apparently increase in relative abundance after death, assessing their functions, and comparing their abundance profiles through postmortem time in two species, mouse and zebrafish. We found mRNA transcript profiles of 1063 genes became significantly more abundant after death of healthy adult animals in a time series spanning up to 96 h postmortem. Ordination plots revealed non-random patterns in the profiles by time. While most of these transcript levels increased within 0.5 h postmortem, some increased only at 24 and 48 h postmortem. Functional characterization of the most abundant transcripts revealed the following categories: stress, immunity, inflammation, apoptosis, transport, development, epigenetic regulation and cancer. The data suggest a step-wise shutdown occurs in organismal death that is manifested by the apparent increase of certain transcripts with various abundance maxima and durations.
Keywords: Gene Meters; cancer; developmental control; forensic science; postmortem transcriptome; transplantology.
© 2017 The Authors.
Figures

References
-
- Monod J. 1961. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 3, 318–356. (doi:10.1016/S0022-2836(61)80072-7) - DOI - PubMed
-
- Zong WX, Thompson CB. 2006. Necrotic death as a cell fate. Genes Dev. 20, 1–15. (doi:10.1101/gad.1376506) - DOI - PubMed
-
- Galluzzi L, Bravo-San Pedro JM, Kroemer G. 2014. Organelle-specific initiation of cell death. Nat. Cell. Biol. 16, 728–736. (doi:10.1038/ncb3005) - DOI - PubMed
-
- Ferri KF, Kroemer G. 2001. Organelle-specific initiation of cell death pathways. Nat. Cell. Biol. 3, E255–E263. (doi:10.1038/ncb1101-e255) - DOI - PubMed
-
- Syntichaki P, Tavernarakis N. 2002. Death by necrosis. Uncontrollable catastrophe, or is there order behind the chaos? EMBO Rep. 3, 604–609. (doi:10.1093/embo-reports/kvf138) - DOI - PMC - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources