

Prokaryotic Heme Biosynthesis: Multiple Pathways to a Common Essential Product
- PMID: 28123057
- PMCID: PMC5312243
- DOI: 10.1128/MMBR.00048-16
Prokaryotic Heme Biosynthesis: Multiple Pathways to a Common Essential Product
Abstract
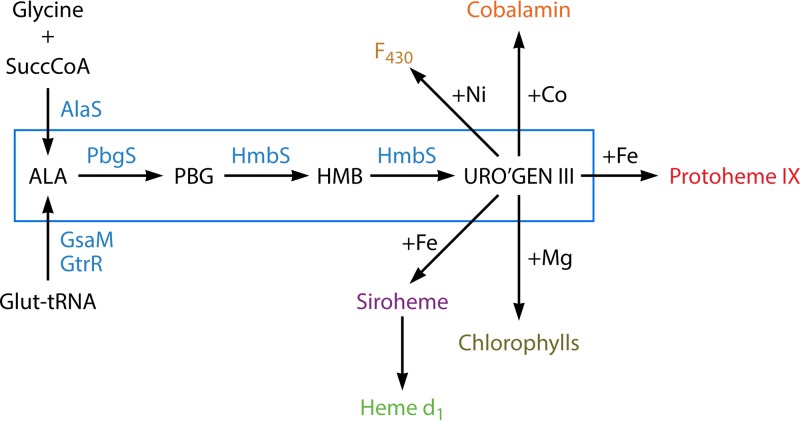
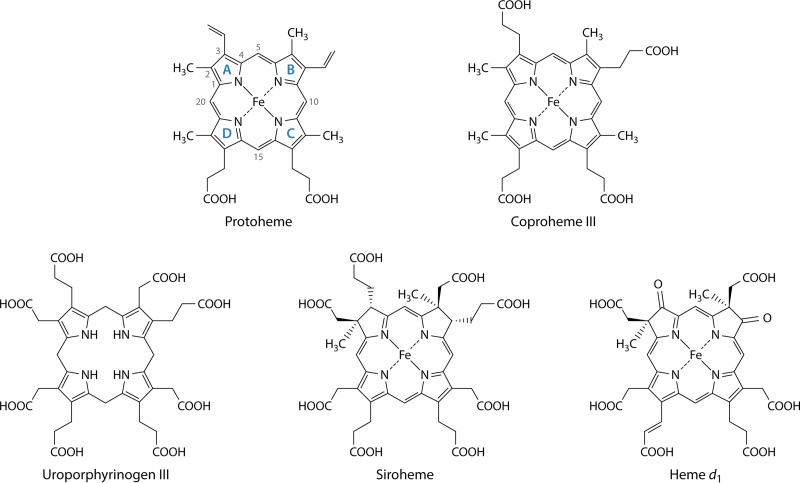
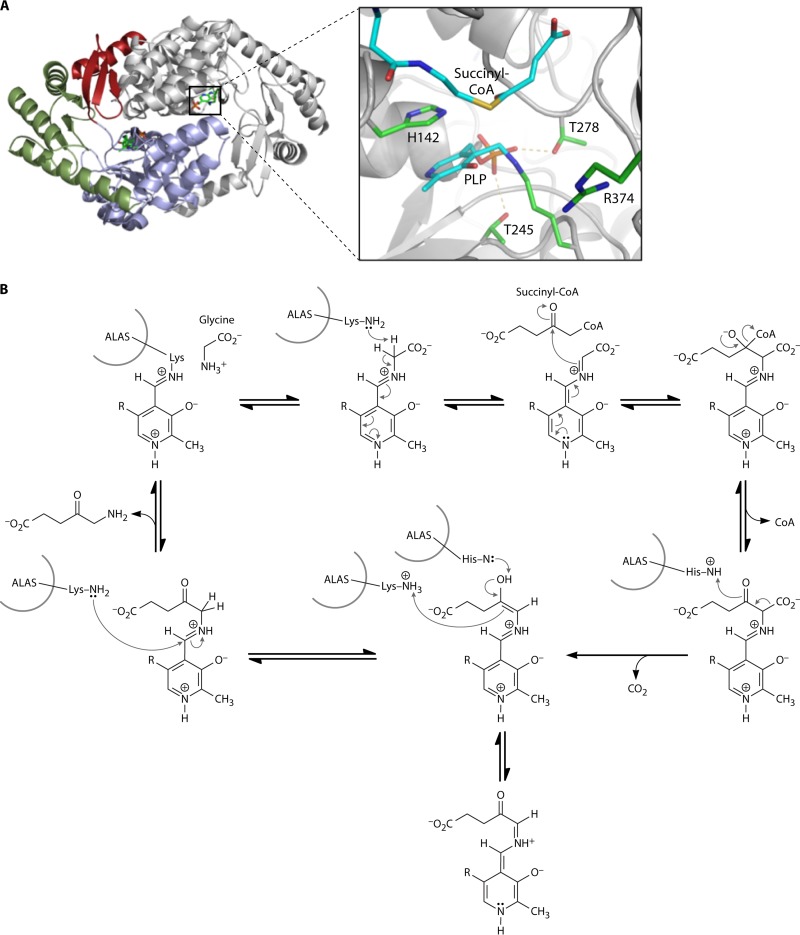
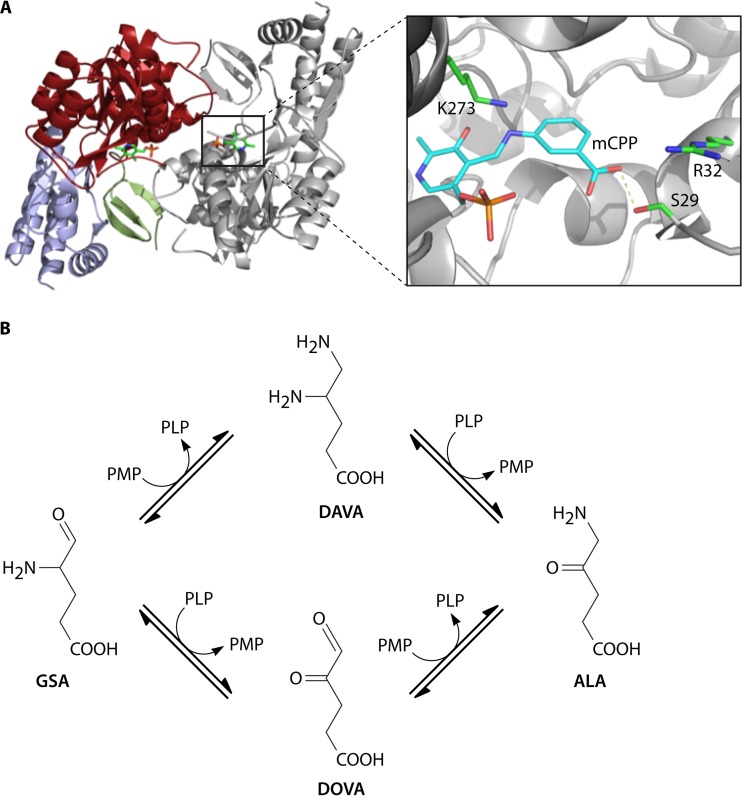
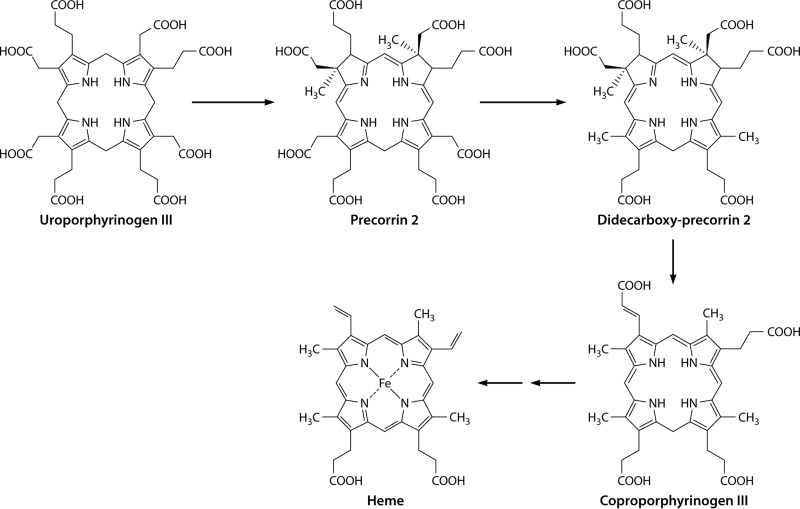
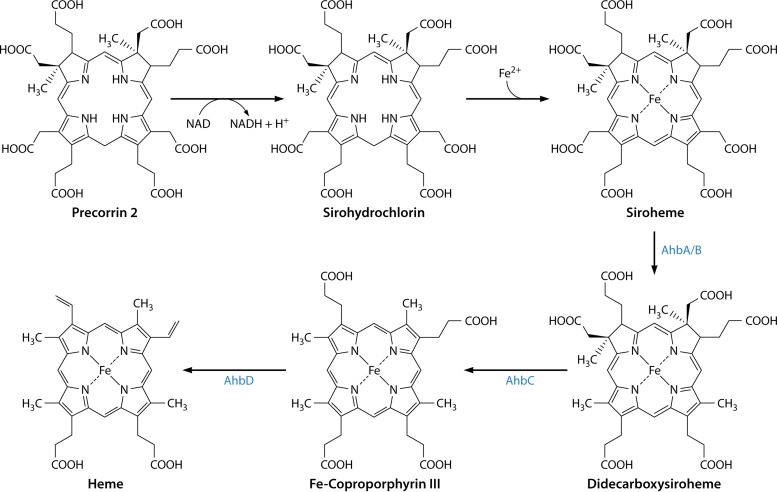
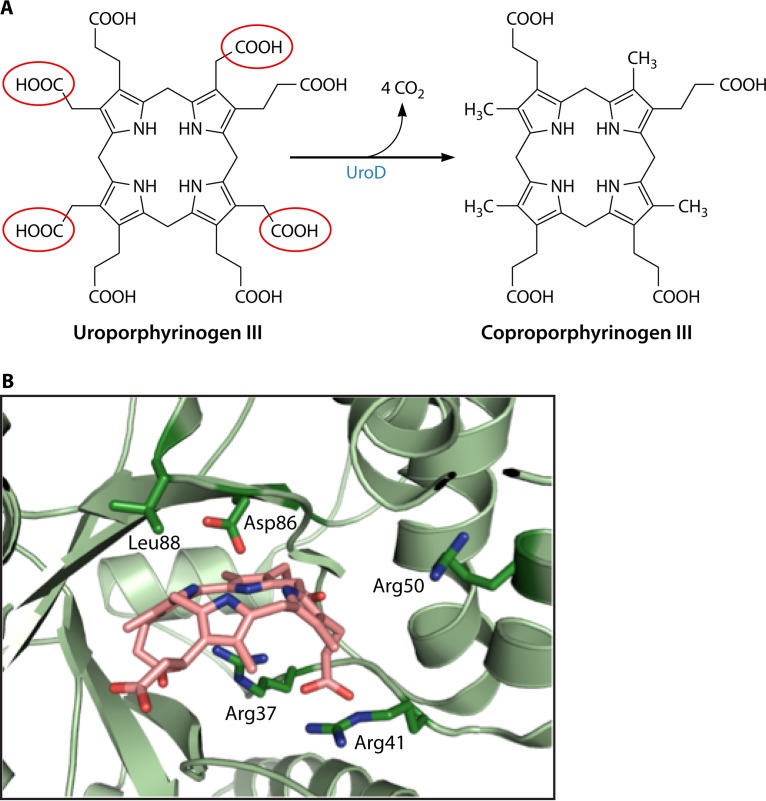
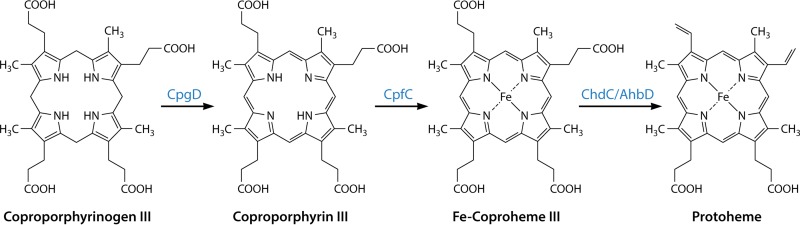
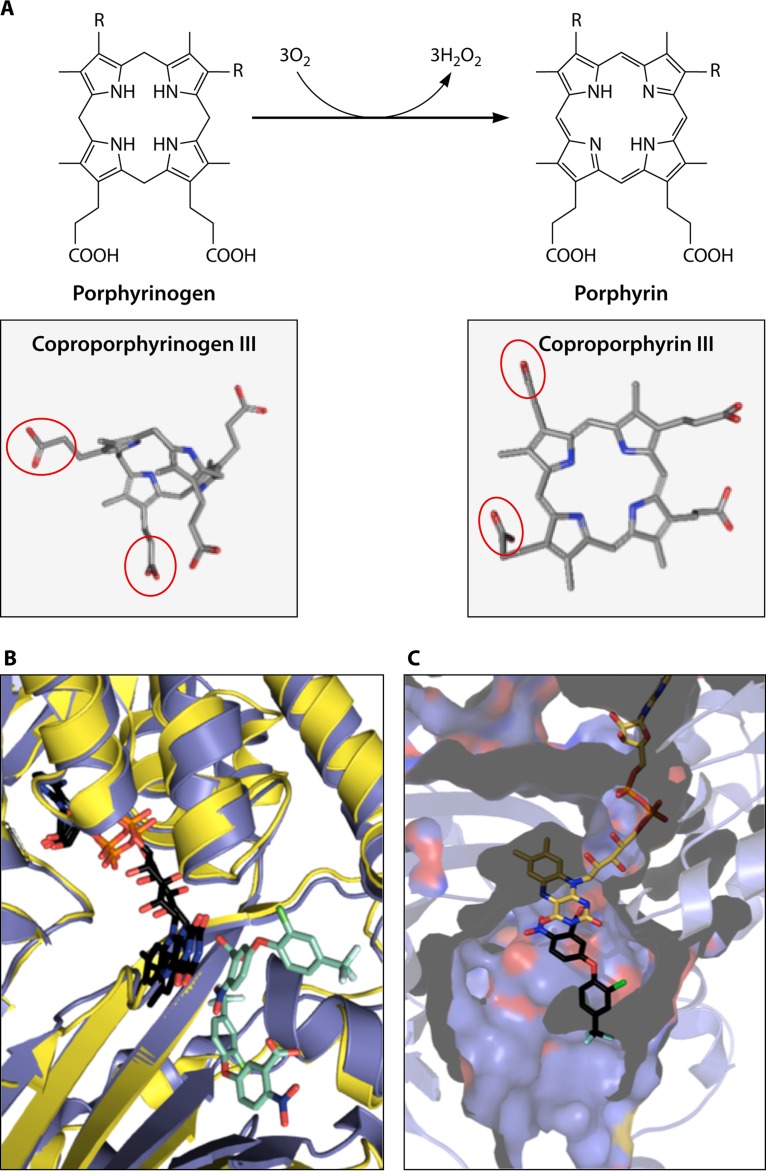
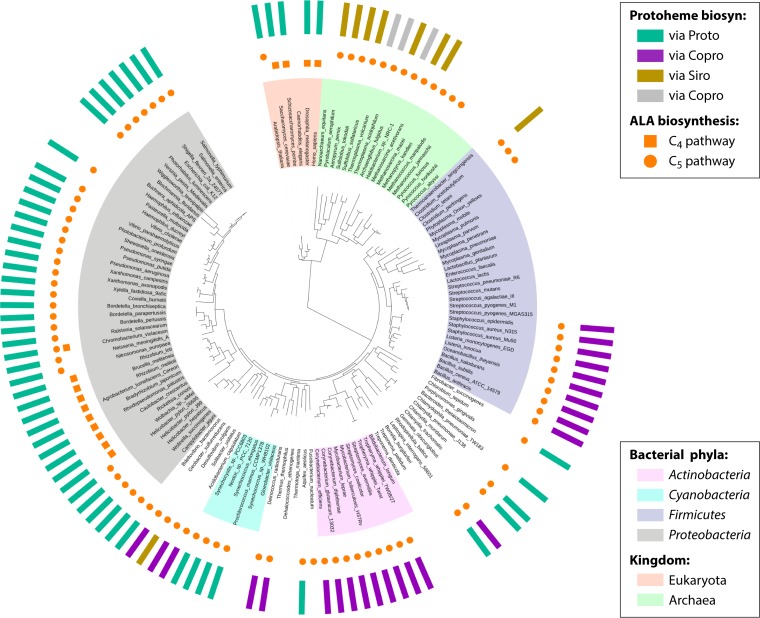
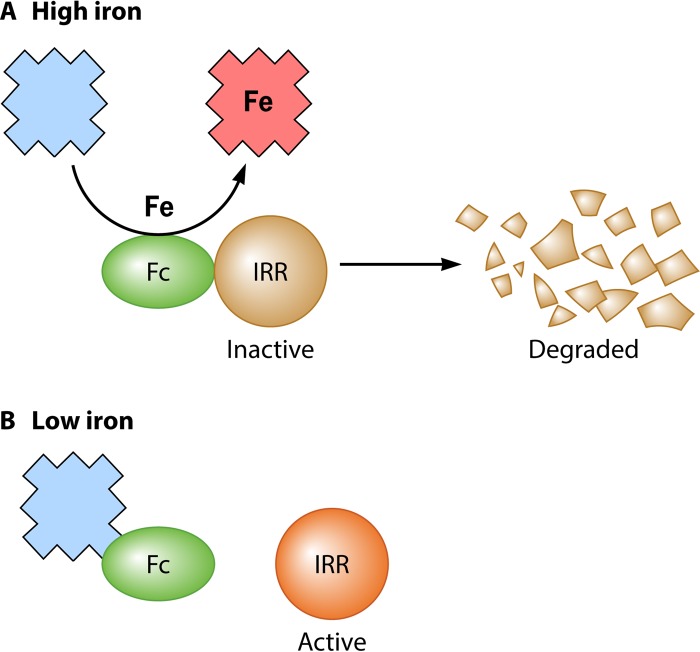
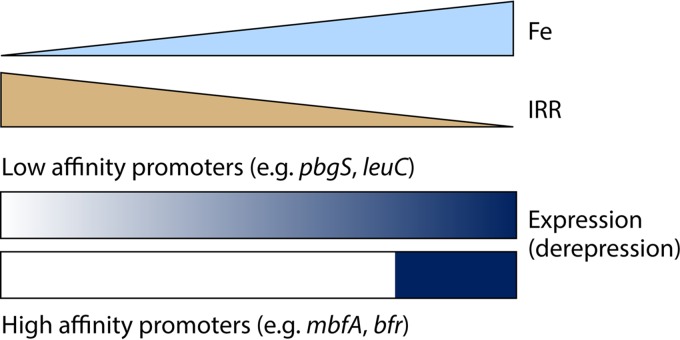
The advent of heme during evolution allowed organisms possessing this compound to safely and efficiently carry out a variety of chemical reactions that otherwise were difficult or impossible. While it was long assumed that a single heme biosynthetic pathway existed in nature, over the past decade, it has become clear that there are three distinct pathways among prokaryotes, although all three pathways utilize a common initial core of three enzymes to produce the intermediate uroporphyrinogen III. The most ancient pathway and the only one found in the Archaea converts siroheme to protoheme via an oxygen-independent four-enzyme-step process. Bacteria utilize the initial core pathway but then add one additional common step to produce coproporphyrinogen III. Following this step, Gram-positive organisms oxidize coproporphyrinogen III to coproporphyrin III, insert iron to make coproheme, and finally decarboxylate coproheme to protoheme, whereas Gram-negative bacteria first decarboxylate coproporphyrinogen III to protoporphyrinogen IX and then oxidize this to protoporphyrin IX prior to metal insertion to make protoheme. In order to adapt to oxygen-deficient conditions, two steps in the bacterial pathways have multiple forms to accommodate oxidative reactions in an anaerobic environment. The regulation of these pathways reflects the diversity of bacterial metabolism. This diversity, along with the late recognition that three pathways exist, has significantly slowed advances in this field such that no single organism's heme synthesis pathway regulation is currently completely characterized.
Keywords: biosynthetic pathways; heme; metabolic regulation; pathway evolution; tetrapyrroles.
Copyright © 2017 American Society for Microbiology.
Figures




Similar articles
-
Noncanonical coproporphyrin-dependent bacterial heme biosynthesis pathway that does not use protoporphyrin.Proc Natl Acad Sci U S A. 2015 Feb 17;112(7):2210-5. doi: 10.1073/pnas.1416285112. Epub 2015 Feb 2. Proc Natl Acad Sci U S A. 2015. PMID: 25646457 Free PMC article.
-
[Unusual pathways and environmentally regulated genes of bacterial heme biosynthesis].Naturwissenschaften. 1996 Sep;83(9):389-400. Naturwissenschaften. 1996. PMID: 8965922 Review. German.
-
Heme biosynthesis in prokaryotes.Biochim Biophys Acta Mol Cell Res. 2021 Jan;1868(1):118861. doi: 10.1016/j.bbamcr.2020.118861. Epub 2020 Sep 23. Biochim Biophys Acta Mol Cell Res. 2021. PMID: 32976912 Review.
-
A primer on heme biosynthesis.Biol Chem. 2022 Aug 29;403(11-12):985-1003. doi: 10.1515/hsz-2022-0205. Print 2022 Nov 25. Biol Chem. 2022. PMID: 36029525 Review.
-
Bacillus subtilis HemY is a peripheral membrane protein essential for protoheme IX synthesis which can oxidize coproporphyrinogen III and protoporphyrinogen IX.J Bacteriol. 1994 Oct;176(19):5962-70. doi: 10.1128/jb.176.19.5962-5970.1994. J Bacteriol. 1994. PMID: 7928957 Free PMC article.
Cited by
-
Pathways of Iron and Sulfur Acquisition, Cofactor Assembly, Destination, and Storage in Diverse Archaeal Methanogens and Alkanotrophs.J Bacteriol. 2021 Aug 9;203(17):e0011721. doi: 10.1128/JB.00117-21. Epub 2021 Aug 9. J Bacteriol. 2021. PMID: 34124941 Free PMC article.
-
Mechanisms of metal-dependent non-redox decarboxylases from quantum chemical calculations.Comput Struct Biotechnol J. 2021 May 26;19:3176-3186. doi: 10.1016/j.csbj.2021.05.044. eCollection 2021. Comput Struct Biotechnol J. 2021. PMID: 34141138 Free PMC article. Review.
-
Maintenance of heme homeostasis in Staphylococcus aureus through post-translational regulation of glutamyl-tRNA reductase.J Bacteriol. 2023 Sep 26;205(9):e0017123. doi: 10.1128/jb.00171-23. Epub 2023 Sep 1. J Bacteriol. 2023. PMID: 37655914 Free PMC article.
-
Fusion/fission protein family identification in Archaea.mSystems. 2024 Jun 18;9(6):e0094823. doi: 10.1128/msystems.00948-23. Epub 2024 May 3. mSystems. 2024. PMID: 38700364 Free PMC article.
-
Protein complex formation during denitrification by Pseudomonas aeruginosa.Microb Biotechnol. 2017 Nov;10(6):1523-1534. doi: 10.1111/1751-7915.12851. Epub 2017 Aug 31. Microb Biotechnol. 2017. PMID: 28857512 Free PMC article. Review.
References
-
- Rinaldo S, Sam KA, Castiglione N, Stelitano V, Arcovito A, Brunori M, Allen JW, Ferguson SJ, Cutruzzola F. 2011. Observation of fast release of NO from ferrous d(1) haem allows formulation of a unified reaction mechanism for cytochrome cd(1) nitrite reductases. Biochem J 435:217–225. doi:10.1042/BJ20101615. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
