

Genome Analysis of Clostridium difficile PCR Ribotype 014 Lineage in Australian Pigs and Humans Reveals a Diverse Genetic Repertoire and Signatures of Long-Range Interspecies Transmission
- PMID: 28123380
- PMCID: PMC5225093
- DOI: 10.3389/fmicb.2016.02138
Genome Analysis of Clostridium difficile PCR Ribotype 014 Lineage in Australian Pigs and Humans Reveals a Diverse Genetic Repertoire and Signatures of Long-Range Interspecies Transmission
Abstract
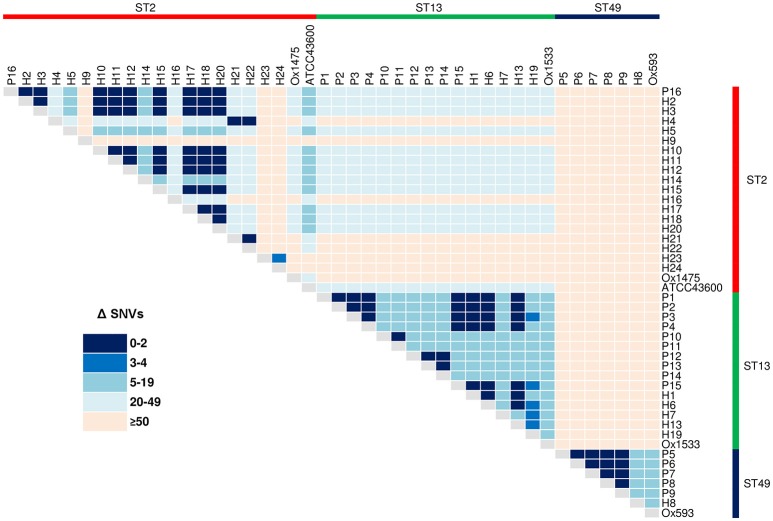
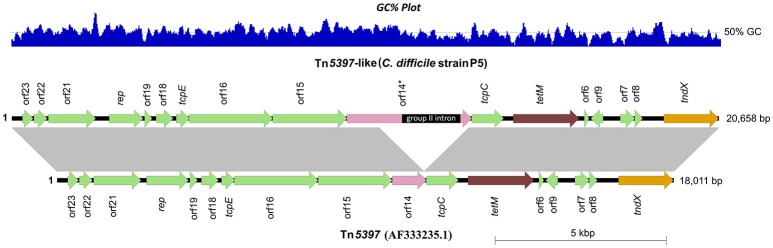
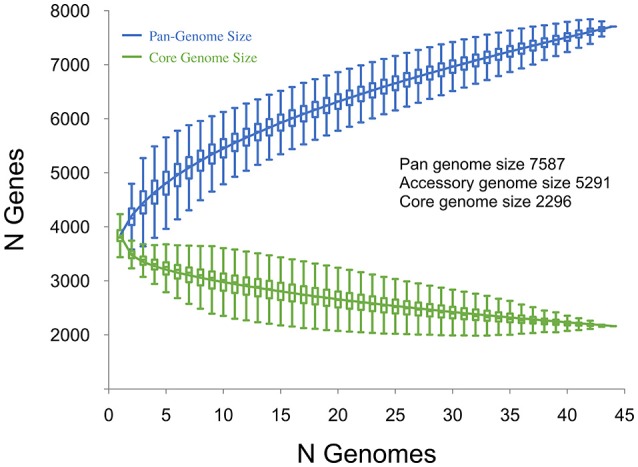
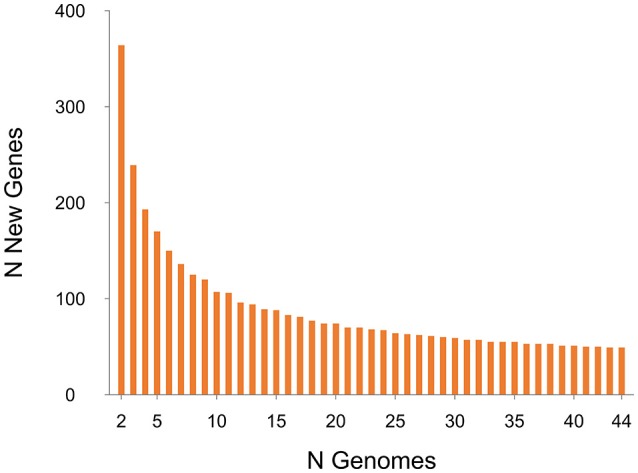
Clostridium difficile PCR ribotype (RT) 014 is well-established in both human and porcine populations in Australia, raising the possibility that C. difficile infection (CDI) may have a zoonotic or foodborne etiology. Here, whole genome sequencing and high-resolution core genome phylogenetics were performed on a contemporaneous collection of 40 Australian RT014 isolates of human and porcine origin. Phylogenies based on MLST (7 loci, STs 2, 13, and 49) and core orthologous genes (1260 loci) showed clustering of human and porcine strains indicative of very recent shared ancestry. Core genome single nucleotide variant (SNV) analysis found 42% of human strains showed a clonal relationship (separated by ≤2 SNVs in their core genome) with one or more porcine strains, consistent with recent inter-host transmission. Clones were spread over a vast geographic area with 50% of the human cases occurring without recent healthcare exposure. These findings suggest a persistent community reservoir with long-range dissemination, potentially due to agricultural recycling of piggery effluent. We also provide the first pan-genome analysis for this lineage, characterizing its resistome, prophage content, and in silico virulence potential. The RT014 is defined by a large "open" pan-genome (7587 genes) comprising a core genome of 2296 genes (30.3% of the total gene repertoire) and an accessory genome of 5291 genes. Antimicrobial resistance genotypes and phenotypes varied across host populations and ST lineages and were characterized by resistance to tetracycline [tetM, tetA(P), tetB(P) and tetW], clindamycin/erythromycin (ermB), and aminoglycosides (aph3-III-Sat4A-ant6-Ia). Resistance was mediated by clinically important mobile genetic elements, most notably Tn6194 (harboring ermB) and a novel variant of Tn5397 (harboring tetM). Numerous clinically important prophages (Siphoviridae and Myoviridae) were identified as well as an uncommon accessory gene regulator locus (agr3). Conservation in the pathogenicity locus and S-layer correlated with ST affiliation, further extending the concept of clonal C. difficile lineages. This study provides novel insights on the genetic variability and strain relatedness of C. difficile RT014, a lineage of emerging One Health importance. Ongoing molecular and genomic surveillance of strains in humans, animals, food, and the environment is imperative to identify opportunities to reduce the overall CDI burden.
Keywords: CDI; One Health; antimicrobial resistance; pan-genome; phylogenomics; porcine; zoonosis.
Figures
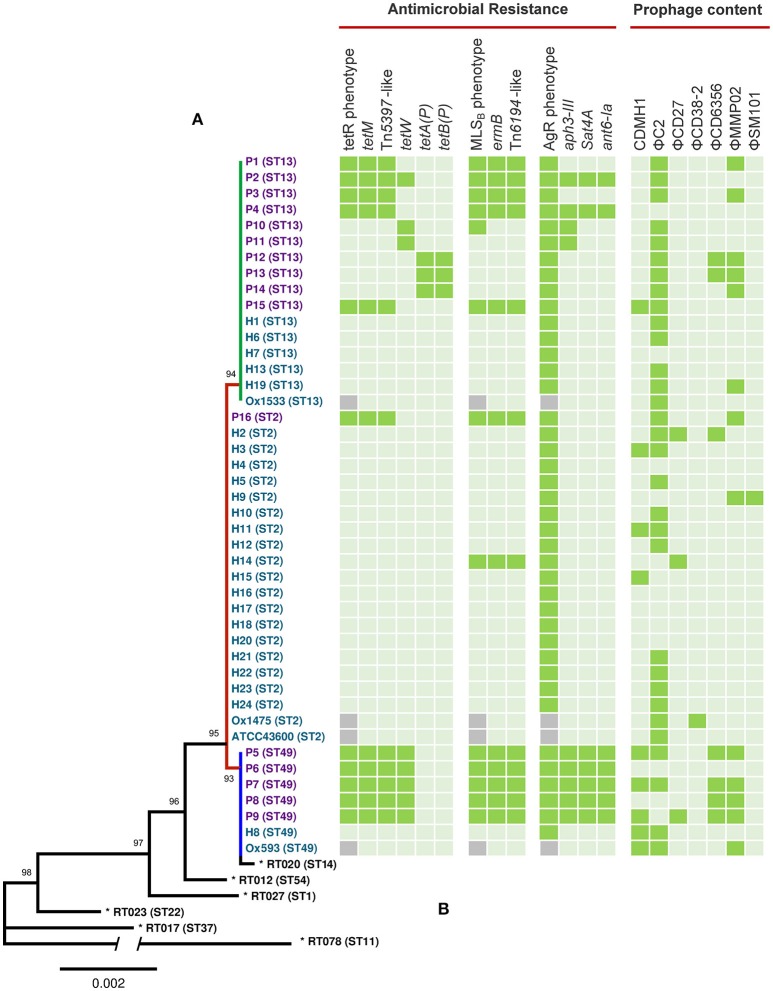
 ), absence (
), absence ( ), MICs were not determined for UK strains Ox1533, Ox1475, Ox1593, and ATCC43600 (
), MICs were not determined for UK strains Ox1533, Ox1475, Ox1593, and ATCC43600 ( ). Some genomes harbored duplicate copies of prophages; P3 (2x ΦC2), P7 (2x ΦC2), P15 (3x ΦC2), H8 (2x ΦC2), H19 (2x ΦC2 and 2x ΦMMP02), and Ox1475 (2x ΦC2).
). Some genomes harbored duplicate copies of prophages; P3 (2x ΦC2), P7 (2x ΦC2), P15 (3x ΦC2), H8 (2x ΦC2), H19 (2x ΦC2 and 2x ΦMMP02), and Ox1475 (2x ΦC2).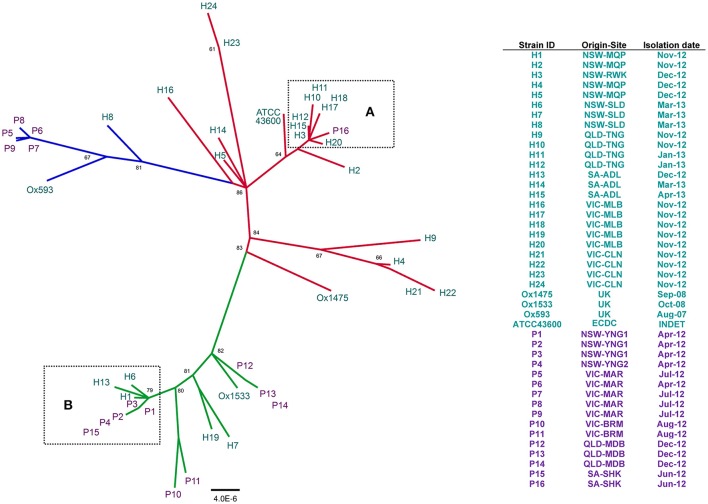
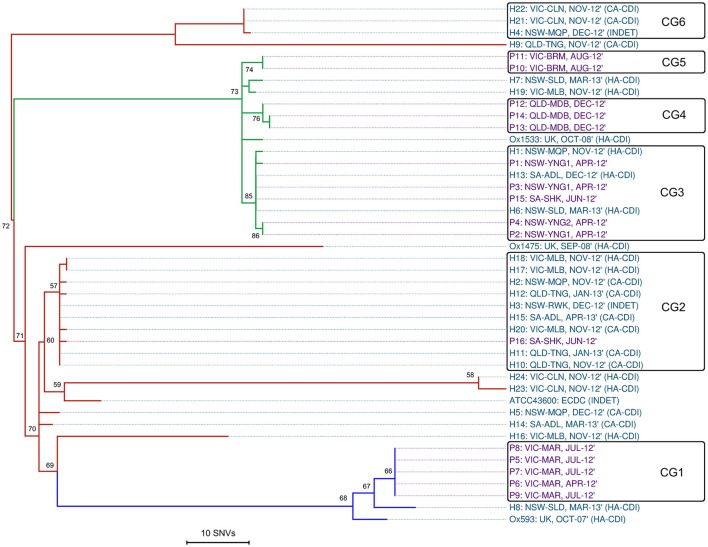

References
-
- ACSQHC (2015). Antimicrobial Prescribing Practice in Australian Hospitals: Results of the 2014 National Antimicrobial Prescribing Survey. Australian Commission on Safety and Quality in Health Care Sydney, NSW. Available online at: http://www.safetyandquality.gov.au/
LinkOut - more resources
Full Text Sources
Other Literature Sources