

Assessing the Genotypic Differences between Strains of Corynebacterium pseudotuberculosis biovar equi through Comparative Genomics
- PMID: 28125655
- PMCID: PMC5268413
- DOI: 10.1371/journal.pone.0170676
Assessing the Genotypic Differences between Strains of Corynebacterium pseudotuberculosis biovar equi through Comparative Genomics
Abstract
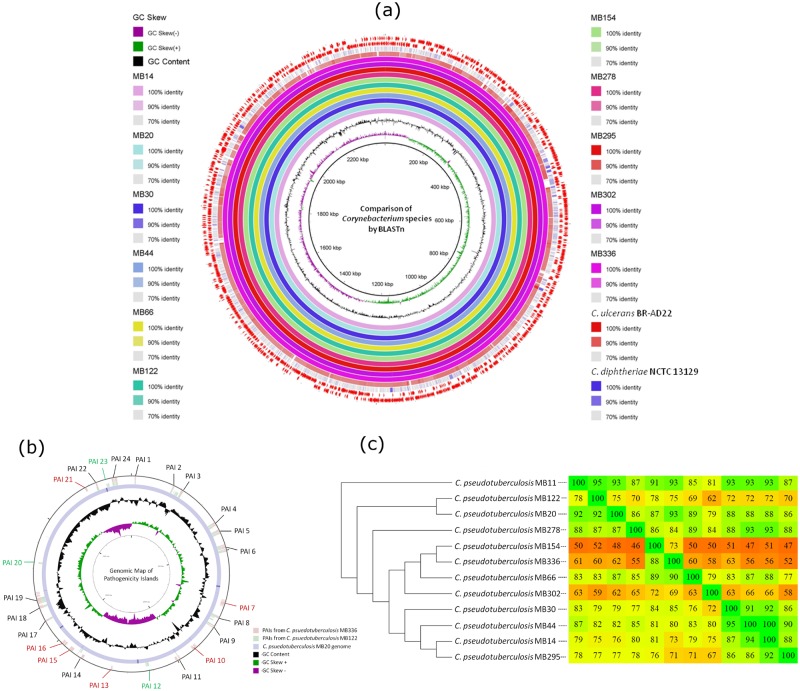
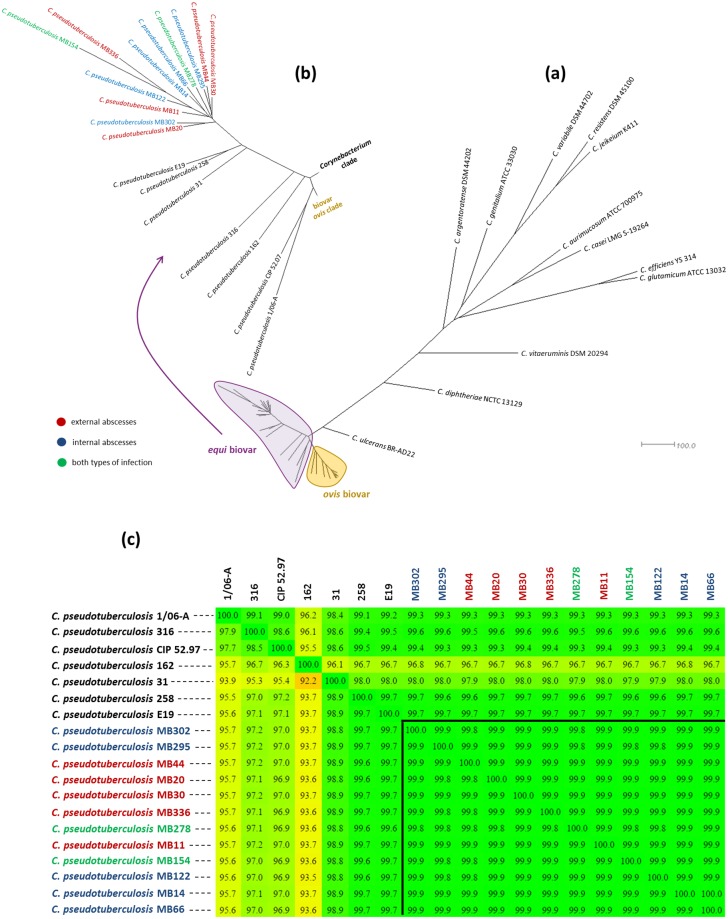
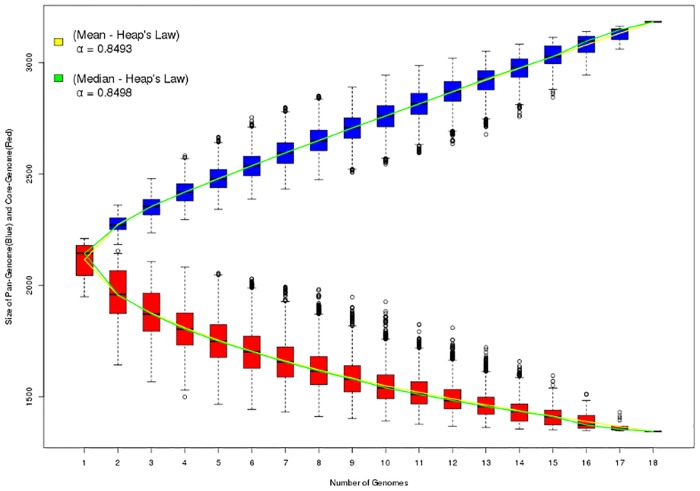
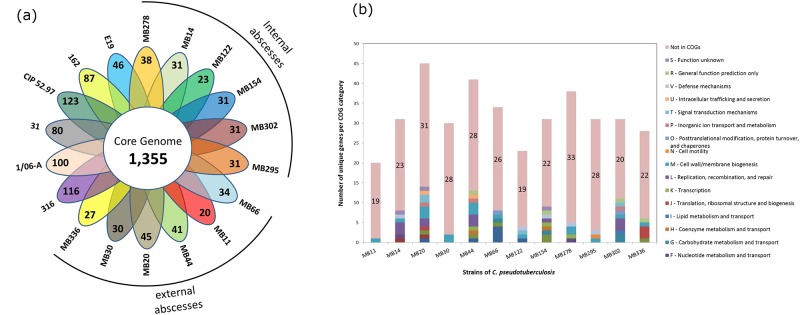
Seven genomes of Corynebacterium pseudotuberculosis biovar equi were sequenced on the Ion Torrent PGM platform, generating high-quality scaffolds over 2.35 Mbp. This bacterium is the causative agent of disease known as "pigeon fever" which commonly affects horses worldwide. The pangenome of biovar equi was calculated and two phylogenomic approaches were used to identify clustering patterns within Corynebacterium genus. Furthermore, other comparative analyses were performed including the prediction of genomic islands and prophages, and SNP-based phylogeny. In the phylogenomic tree, C. pseudotuberculosis was divided into two distinct clades, one formed by nitrate non-reducing species (biovar ovis) and another formed by nitrate-reducing species (biovar equi). In the latter group, the strains isolated from California were more related to each other, while the strains CIP 52.97 and 1/06-A formed the outermost clade of the biovar equi. A total of 1,355 core genes were identified, corresponding to 42.5% of the pangenome. This pangenome has one of the smallest core genomes described in the literature, suggesting a high genetic variability of biovar equi of C. pseudotuberculosis. The analysis of the similarity between the resistance islands identified a higher proximity between the strains that caused more severe infectious conditions (infection in the internal organs). Pathogenicity islands were largely conserved between strains. Several genes that modulate the pathogenicity of C. pseudotuberculosis were described including peptidases, recombination enzymes, micoside synthesis enzymes, bacteriocins with antimicrobial activity and several others. Finally, no genotypic differences were observed between the strains that caused the three different types of infection (external abscess formation, infection with abscess formation in the internal organs, and ulcerative lymphangitis). Instead, it was noted that there is a higher phenetic correlation between strains isolated at California compared to the other strains. Additionally, high variability of resistance islands suggests gene acquisition through several events of horizontal gene transfer.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
The pan-genome of the animal pathogen Corynebacterium pseudotuberculosis reveals differences in genome plasticity between the biovar ovis and equi strains.PLoS One. 2013;8(1):e53818. doi: 10.1371/journal.pone.0053818. Epub 2013 Jan 14. PLoS One. 2013. PMID: 23342011 Free PMC article.
-
Molecular epidemiology of Corynebacterium pseudotuberculosis isolated from horses in California.Infect Genet Evol. 2017 Apr;49:186-194. doi: 10.1016/j.meegid.2016.12.011. Epub 2016 Dec 13. Infect Genet Evol. 2017. PMID: 27979735
-
A comparative pan-genomic analysis of 53 C. pseudotuberculosis strains based on functional domains.J Biomol Struct Dyn. 2021 Nov;39(18):6974-6986. doi: 10.1080/07391102.2020.1805017. Epub 2020 Aug 11. J Biomol Struct Dyn. 2021. PMID: 32779519
-
Molecular and infection biology of the horse pathogen Rhodococcus equi.FEMS Microbiol Rev. 2009 Sep;33(5):870-91. doi: 10.1111/j.1574-6976.2009.00181.x. Epub 2009 Apr 23. FEMS Microbiol Rev. 2009. PMID: 19453748 Review.
-
A description of genes of Corynebacterium pseudotuberculosis useful in diagnostics and vaccine applications.Genet Mol Res. 2008 Mar 18;7(1):252-60. doi: 10.4238/vol7-1gmr438. Genet Mol Res. 2008. PMID: 18551390 Review.
Cited by
-
Corynebacterium pseudotuberculosis: Whole genome sequencing reveals unforeseen and relevant genetic diversity in this pathogen.PLoS One. 2024 Aug 26;19(8):e0309282. doi: 10.1371/journal.pone.0309282. eCollection 2024. PLoS One. 2024. PMID: 39186721 Free PMC article.
-
Within-Species Genomic Variation and Variable Patterns of Recombination in the Tetracycline Producer Streptomyces rimosus.Front Microbiol. 2019 Mar 21;10:552. doi: 10.3389/fmicb.2019.00552. eCollection 2019. Front Microbiol. 2019. PMID: 30949149 Free PMC article.
-
Isolation, characterization, and pathogenicity assessment of Corynebacterium pseudotuberculosis biovar equi strains from alpacas (Vicugna pacos) in China.Front Microbiol. 2023 Jul 3;14:1206187. doi: 10.3389/fmicb.2023.1206187. eCollection 2023. Front Microbiol. 2023. PMID: 37465023 Free PMC article.
-
Lactobacillus acidophilus protects against Corynebacterium pseudotuberculosis infection by regulating the autophagy of macrophages and maintaining gut microbiota homeostasis in C57BL/6 mice.mSystems. 2024 Jul 23;9(7):e0048424. doi: 10.1128/msystems.00484-24. Epub 2024 Jun 27. mSystems. 2024. PMID: 38934644 Free PMC article.
-
Genotypic differences between strains of the opportunistic pathogen Corynebacterium bovis isolated from humans, cows, and rodents.PLoS One. 2018 Dec 26;13(12):e0209231. doi: 10.1371/journal.pone.0209231. eCollection 2018. PLoS One. 2018. PMID: 30586440 Free PMC article.
References
-
- Aleman M, Spier SJ, Wilson WD, Doherr M. Retrospective study of Corynebacterium pseudotuberculosis infection in horses: 538 cases. J Am Vet Med Ass. 1996; 209: 804–809. - PubMed
-
- Spier SJ, Toth B, Edman J, Quave A, Habasha F, Garrick M, et al. Survival of Corynebacterium pseudotuberculosis biovar equi in soil. Vet Rec. 2012; 170(7): 180. - PubMed
-
- Spier SJ, Leutenegger CM, Carroll SP, Loye JE, Pusterla JB, Carpenter TE, et al. Use of a real-time polymerase chain reaction-based fluorogenic 5’ nuclease assay to evaluate insect vectors of Corynebacterium pseudotuberculosis infections in horses. Am J Vet Res. 2004; 65: 829–834. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
