

Prophylactic versus Therapeutic Fingolimod: Restoration of Presynaptic Defects in Mice Suffering from Experimental Autoimmune Encephalomyelitis
- PMID: 28125677
- PMCID: PMC5268435
- DOI: 10.1371/journal.pone.0170825
Prophylactic versus Therapeutic Fingolimod: Restoration of Presynaptic Defects in Mice Suffering from Experimental Autoimmune Encephalomyelitis
Erratum in
-
Correction: Prophylactic versus Therapeutic Fingolimod: Restoration of Presynaptic Defects in Mice Suffering from Experimental Autoimmune Encephalomyelitis.PLoS One. 2023 Oct 3;18(10):e0292584. doi: 10.1371/journal.pone.0292584. eCollection 2023. PLoS One. 2023. PMID: 37788283 Free PMC article.
Abstract
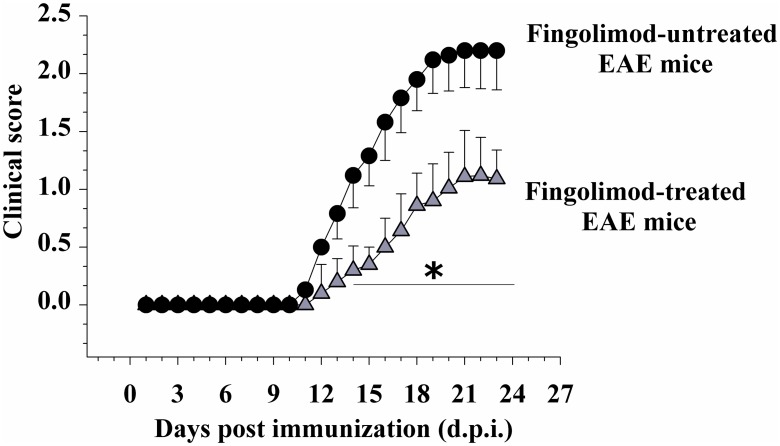
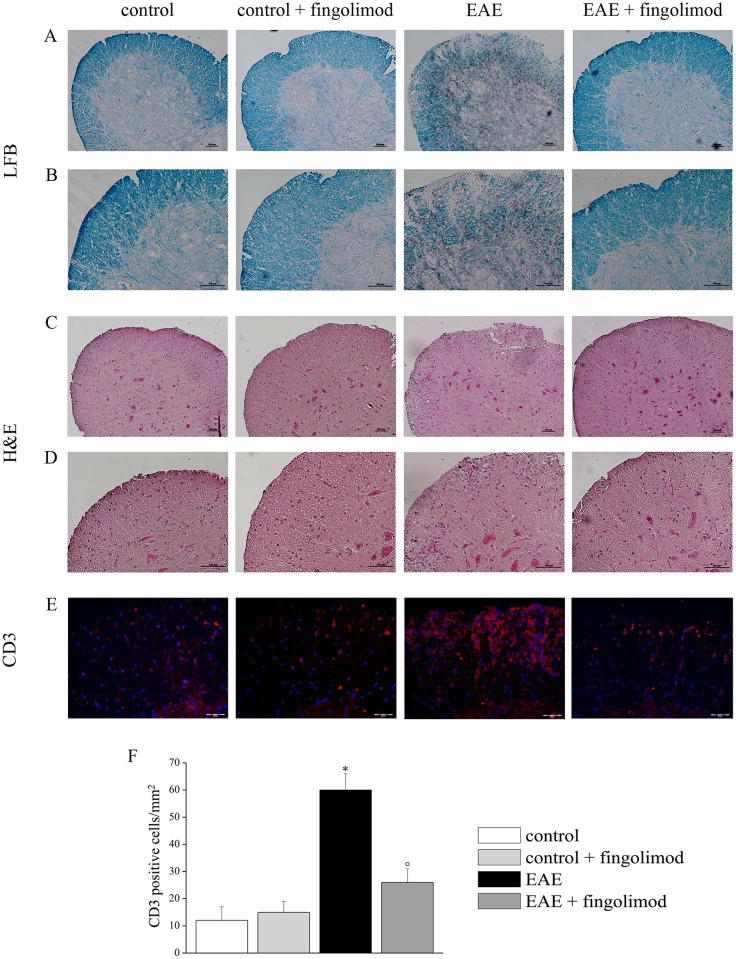
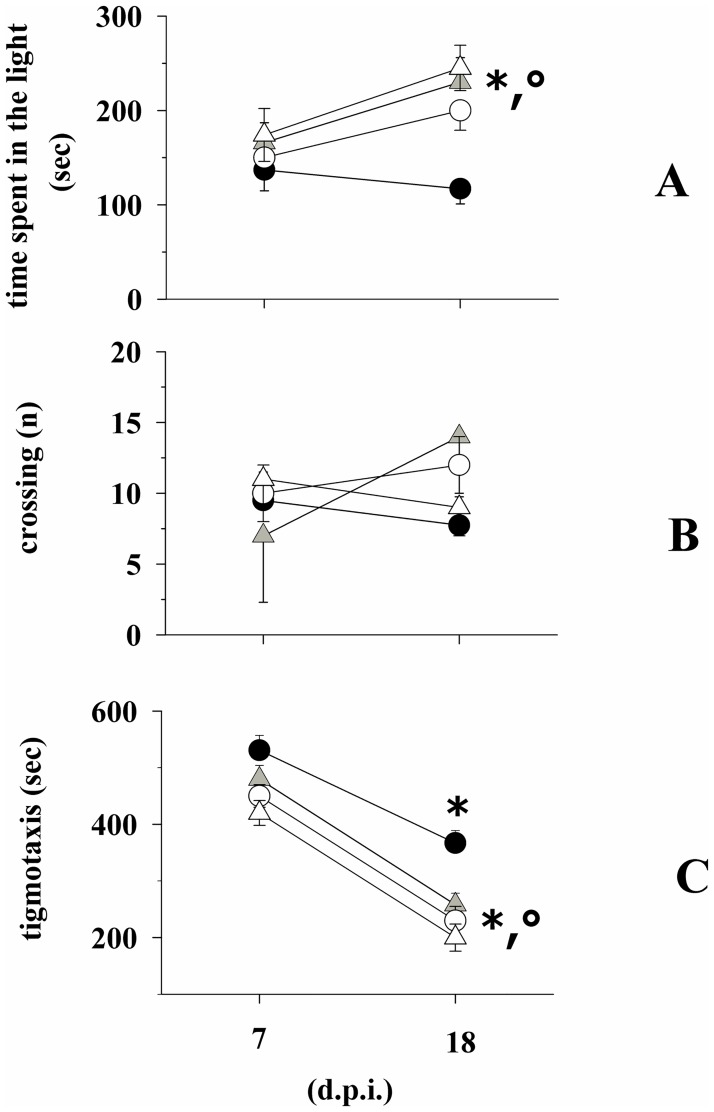
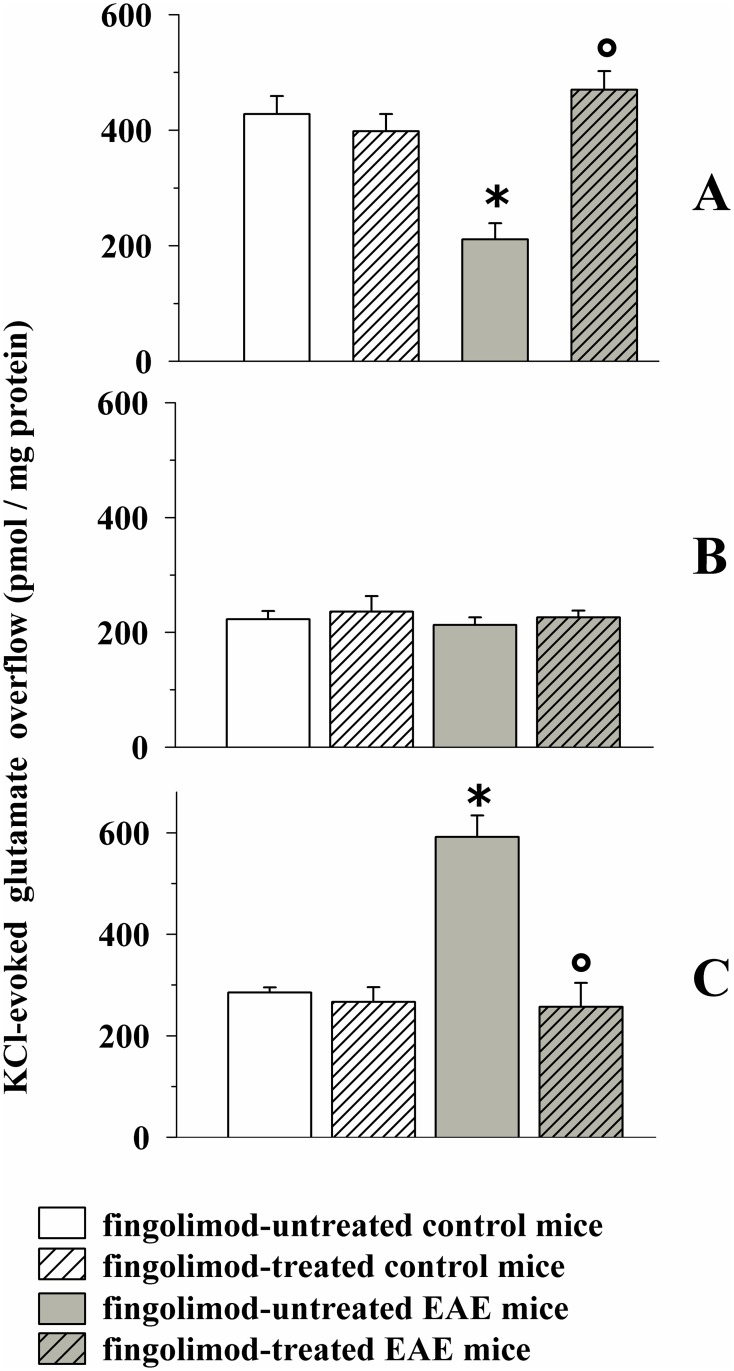
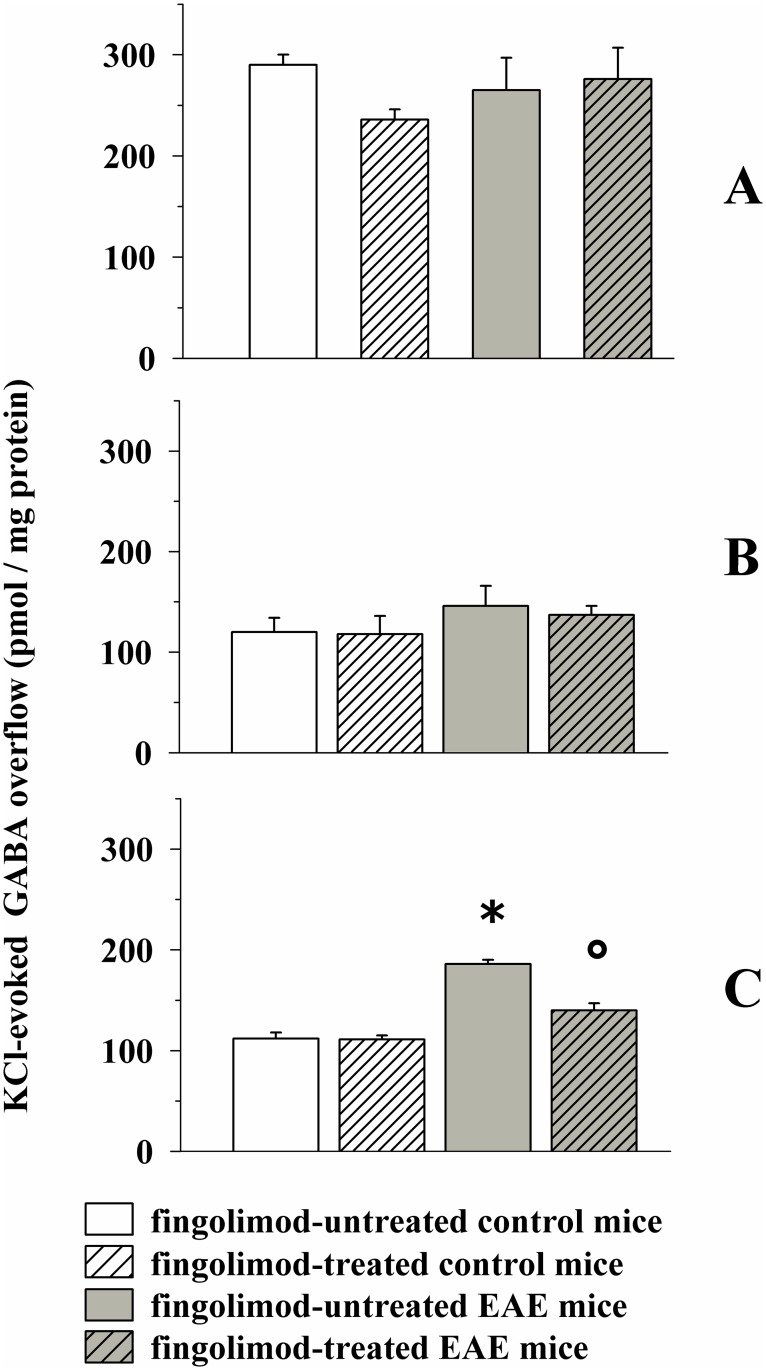
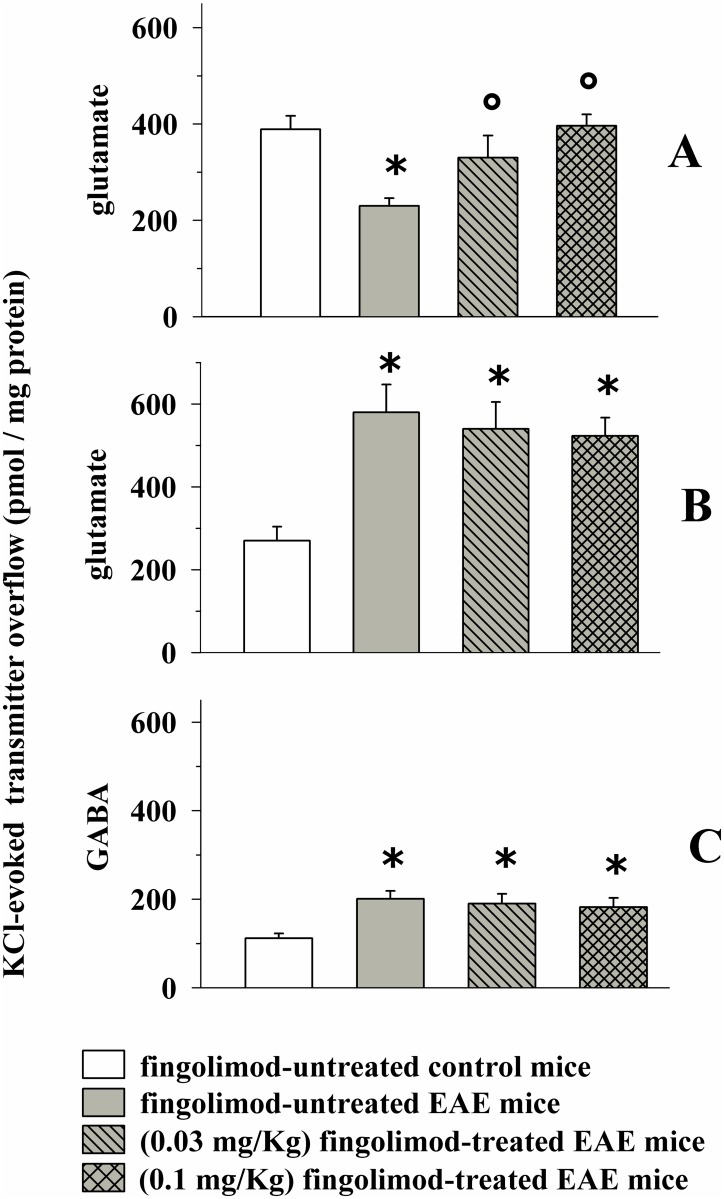
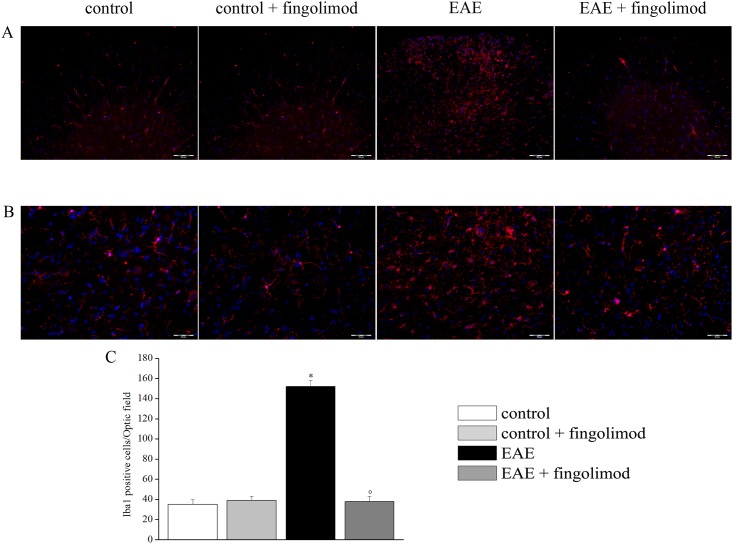
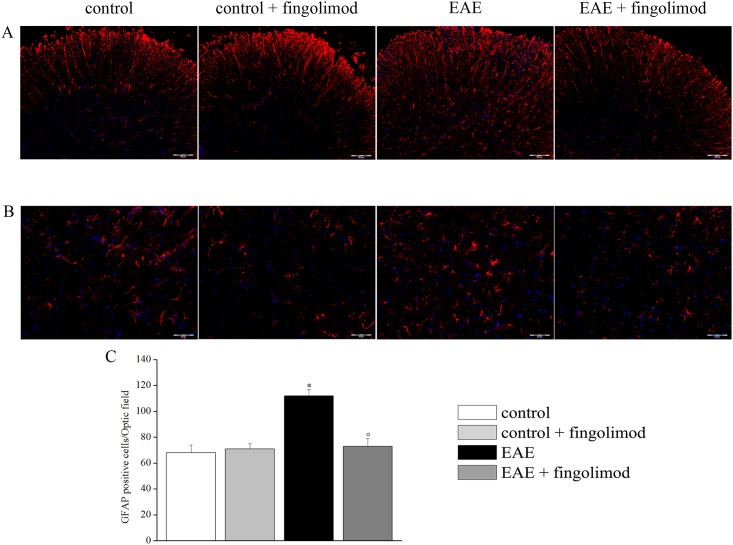
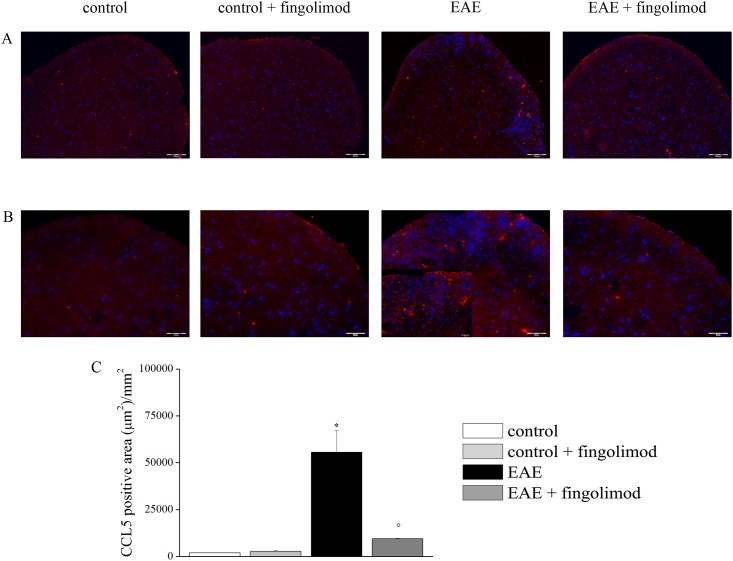
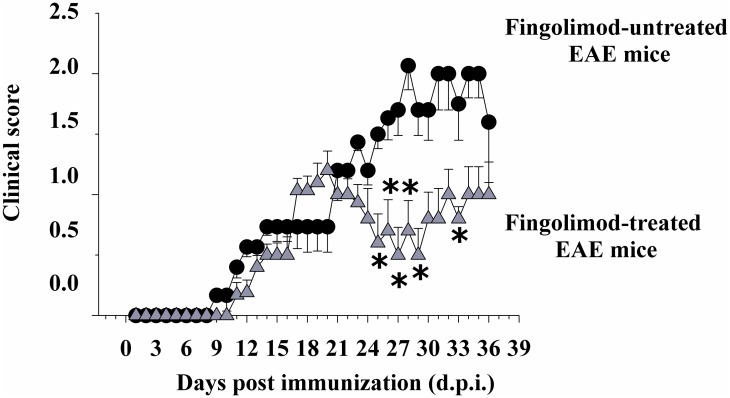
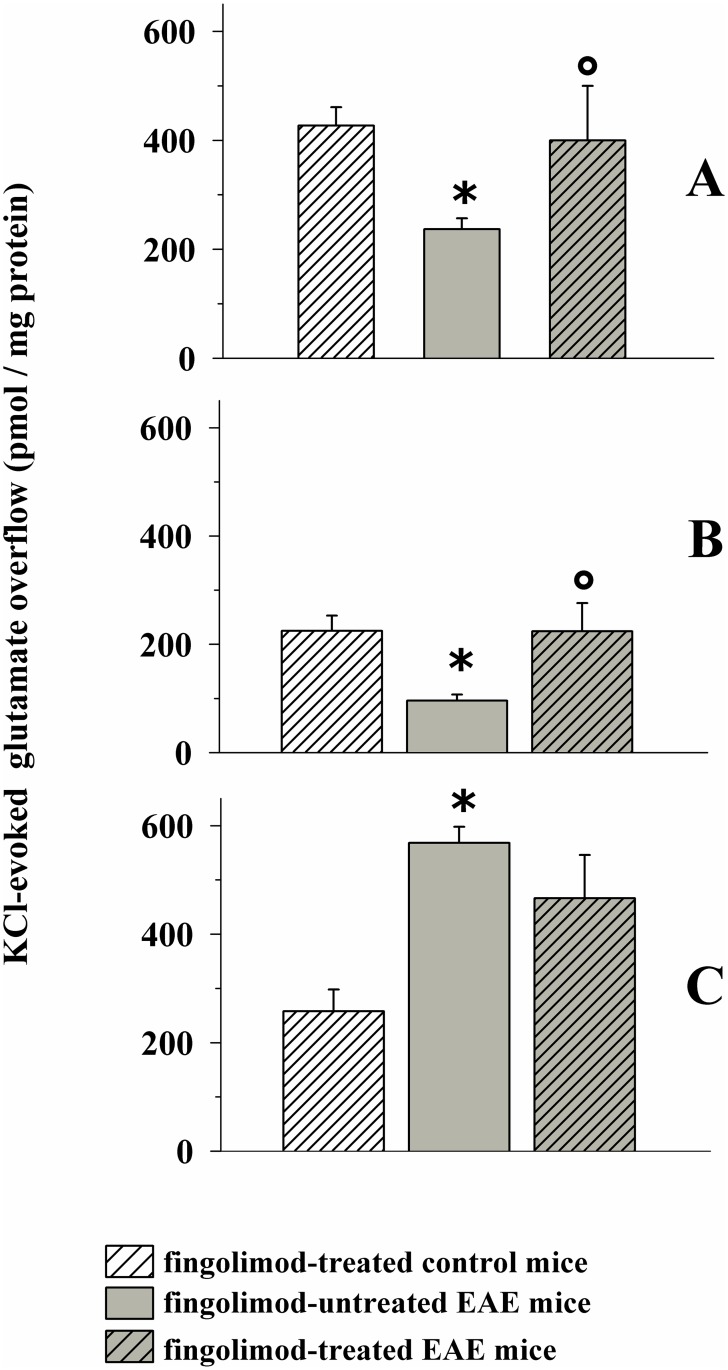
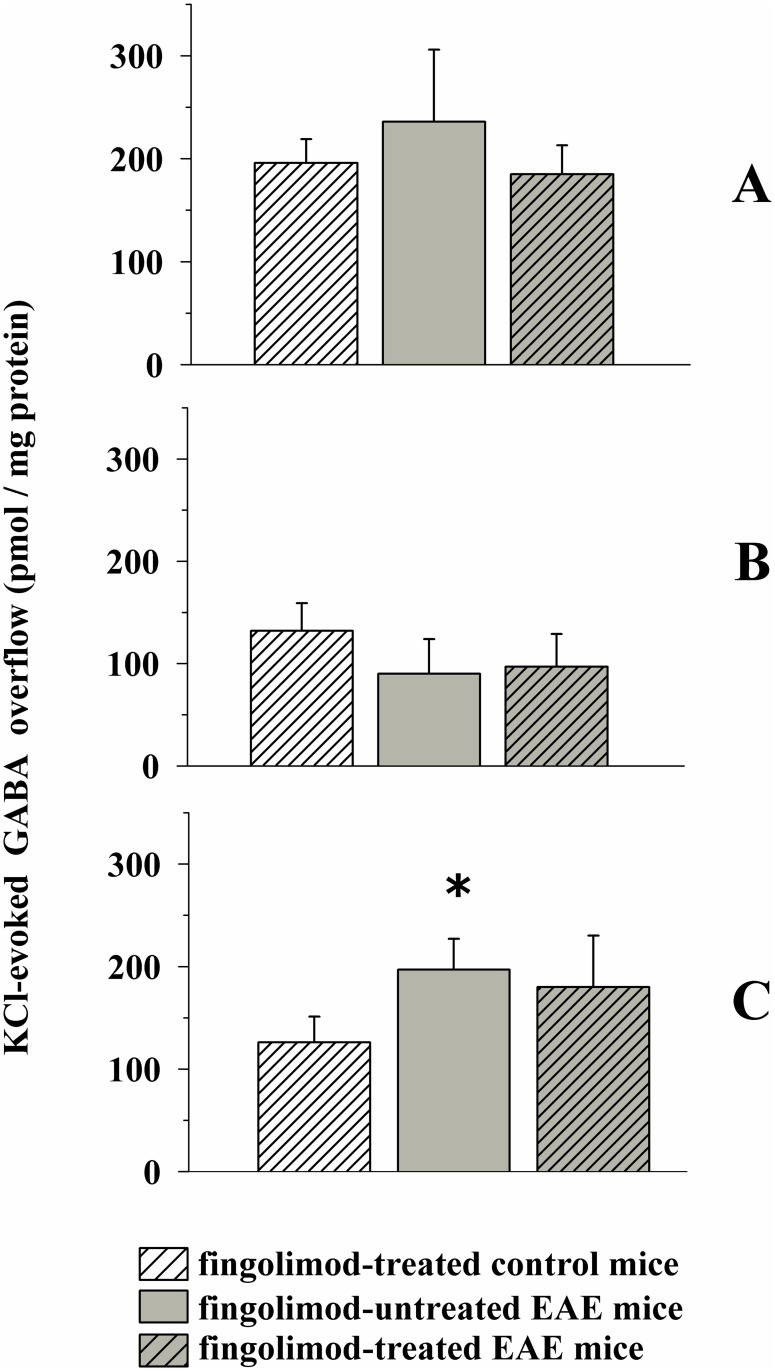
Fingolimod, the first oral, disease-modifying therapy for MS, has been recently proposed to modulate glutamate transmission in the central nervous system (CNS) of mice suffering from Experimental Autoimmune Encephalomyelitis (EAE) and in MS patients. Our study aims at investigating whether oral fingolimod recovers presynaptic defects that occur at different stages of disease in the CNS of EAE mice. In vivo prophylactic (0.3 mg/kg for 14 days, from the 7th day post immunization, d.p.i, the drug dissolved in the drinking water) fingolimod significantly reduced the clinical symptoms and the anxiety-related behaviour in EAE mice. Spinal cord inflammation, demyelination and glial cell activation are markers of EAE progression. These signs were ameliorated following oral fingolimod administration. Glutamate exocytosis was shown to be impaired in cortical and spinal cord terminals isolated from EAE mice at 21 ± 1 d.p.i., while GABA alteration emerged only at the spinal cord level. Prophylactic fingolimod recovered these presynaptic defects, restoring altered glutamate and GABA release efficiency. The beneficial effect occurred in a dose-dependent, region-specific manner, since lower (0.1-0.03 mg/kg) doses restored, although to a different extent, synaptic defects in cortical but not spinal cord terminals. A delayed reduction of glutamate, but not of GABA, exocytosis was observed in hippocampal terminals of EAE mice at 35 d.p.i. Therapeutic (0.3 mg/kg, from 21 d.p.i. for 14 days) fingolimod restored glutamate exocytosis in the cortex and in the hippocampus of EAE mice at 35 ± 1 d.p.i. but not in the spinal cord, where also GABAergic defects remained unmodified. These results improve our knowledge of the molecular events accounting for the beneficial effects elicited by fingolimod in demyelinating disorders.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
