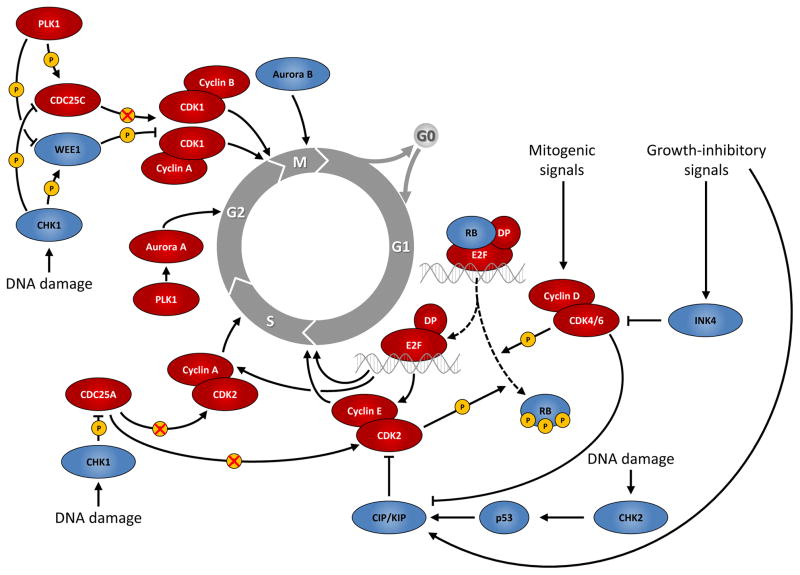
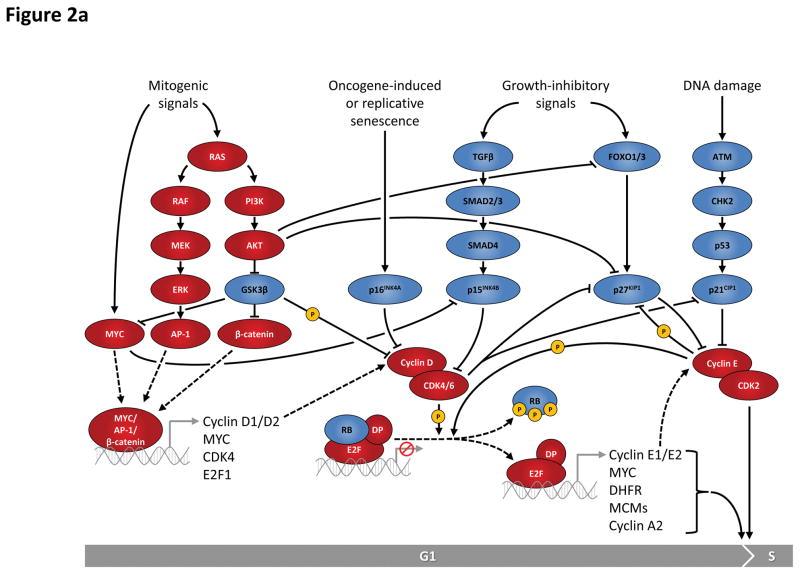
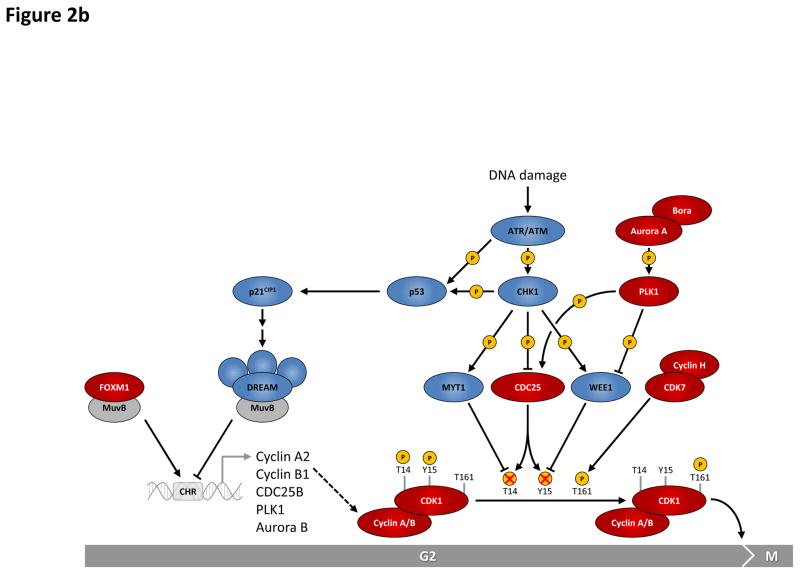
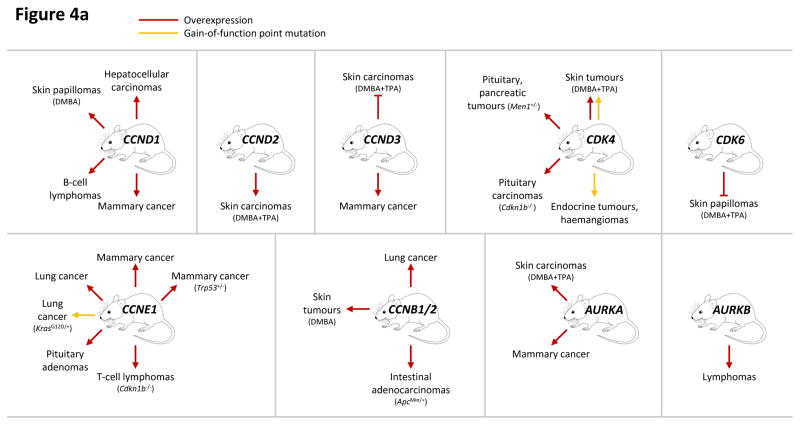
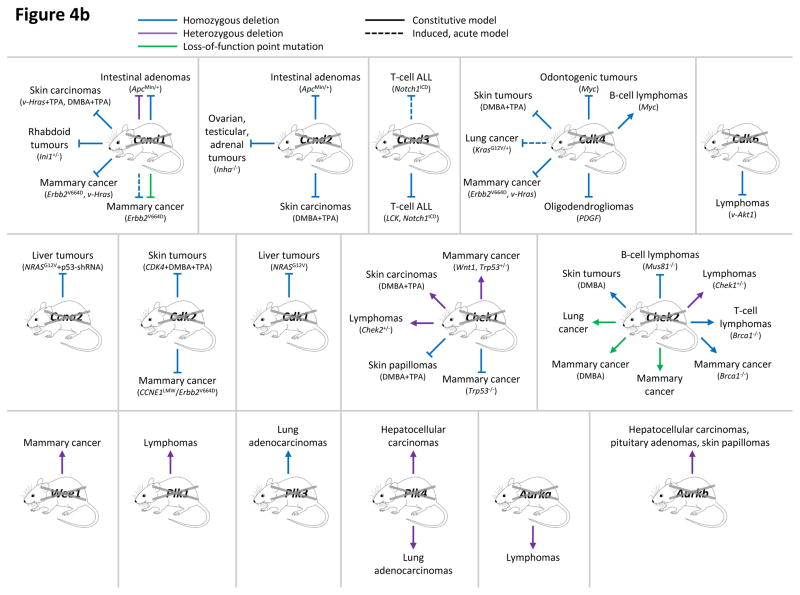
This figure summarizes genetic mouse models used to investigate the role of cell cycle proteins in tumorigenesis. In case of transgenic overexpression, enhanced cancer formation is depicted by red arrows, inhibition of tumorigenesis by red inhibition symbols. Orange arrows indicate cancer formation induced by gain-of-function point mutations. In case of loss-of-function mutations (depicted by crossed out gene symbols), blue inhibition symbols indicate that homozygous ablation of a given gene prevented tumorigenesis, thereby revealing the requirement for this cell cycle protein in cancer formation. Blue dashed inhibition symbols depict an inducible, acute shutdown of
Ccnd1,
Ccnd3 or
Cdk4, used to demonstrate a critical role for these proteins in tumour progression. Arrows indicate that homozygous (blue) or heterozygous (violet) deletion of a cell cycle gene accelerated tumorigenesis. In case of loss-of-function point mutations (as opposed to gene inactivation by deletion described above), enhanced cancer formation is depicted by green arrows, suppressed cancer formation by green inhibition symbols. For tumours induced by a cooperating event (i.e. overexpression or mutation of oncogenes, loss of tumour suppressors or carcinogen treatment), this cooperating event is indicated in parentheses.
a: Genetic mouse models with increased activity of cell cycle proteins, i.e. cyclin D1 (
CCND1), D2 (
CCND2), D3 (
CCND3), CDK4, CDK6, cyclin E1 (
CCNE1), cyclin B1 or B2 (
CCNB1/2), Aurora A (
AURKA) and Aurora B (
AURKB).
b: Genetic mouse models with reduced or abolished activity of cell cycle proteins, i.e. cyclin D1 (
Ccnd1), D2 (
Ccnd2), D3 (
Ccnd3), CDK4, CDK6, cyclin A2 (
Ccna2), CDK2, CDK1, checkpoint kinase 1 (
Chek1) and 2 (
Chek2), WEE1, Polo-like kinases 1 (
Plk1), 3 (
Plk3) and 4 (
Plk4), Aurora A (
Aurka) and Aurora B (
Aurka). AKT1, thymoma viral proto-oncogene 1; ALL, acute lymphoblastic leukaemia; APC, adenomatosis polyposis coli; BRCA1, breast cancer 1; CDKN1B, CDK inhibitor 1b (p27
KIP1); DMBA, 7,12-Dimethylbenz[a]anthracene (a carcinogen); ERBB2 (HER2), erb-b2 receptor tyrosine kinase 2; HRAS, Harvey rat sarcoma virus oncogene; INHA, Inhibin alpha; INI1 (SMARCB1), SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1; KRAS, Kirsten rat sarcoma viral oncogene homolog; LCK, lymphocyte protein tyrosine kinase; MEN1, multiple endocrine neoplasia 1; MUS81, MUS81 endonuclease homolog; MYC, myelocytomatosis oncogene; NRAS, neuroblastoma ras oncogene; p53-shRNA, short hairpin RNA targeting p53; PDGFB, platelet derived growth factor, B polypeptide; TPA, 12-O-Tetradecanoylphorbol-13-acetate (a tumour promoter); TRP53, transformation related protein 53 (p53); WNT1, wingless-type MMTV integration site family, member 1.
| CCND1 gain: | Hepatocellular carcinomas; Skin papillomas (DMBA); B-cell lymphomas; Mammary cancer |
| CCND2 gain: | Skin carcinomas (DMBA+TPA) |
| CCND3 gain: | Skin carcinomas (DMBA+TPA); Mammary cancer |
| CDK4 gain: | Skin tumours (DMBA+TPA), ; Pituitary, pancreatic tumours (Men1+/−); Pituitary carcinomas (Cdkn1b−/−); Endocrine tumours, haemangiomas |
| CDK6 gain: | Skin papillomas (DMBA+TPA) |
| CCNE1 gain: | Mammary cancer (Trp53+/−); Mammary cancer; Lung cancer; Lung cancer (KrasG12D/+); Pituitary adenomas; T-cell lymphomas (Cdkn1b−/−) |
| CCNB1/2 gain: | Lung cancer; Skin tumours (DMBA); Intestinal adenocarcinomas (ApcMin/+) |
| AURKA gain: | Skin carcinomas (DMBA+TPA); Mammary cancer |
| AURKB gain: | Lymphomas |
| Ccnd1 loss: | Intestinal adenomas (ApcMin/+); Skin carcinomas (v-Hras+TPA, DMBA+TPA); Rhabdoid tumours (Ini1+/−); Mammary cancer (Erbb2V664D, v-Hras), ; Mammary cancer (Erbb2V664D), |
| Ccnd2 loss: | Intestinal adenomas (ApcMin/+); Ovarian, testicular, adrenal tumours (Inha−/−); Skin carcinomas (DMBA+TPA) |
| Ccnd3 loss: | T-cell ALL (Notch1ICD); T-cell ALL (LCK, Notch1ICD) |
| Cdk4 loss: | Odontogenic tumours (Myc); Skin tumours (DMBA+TPA); Lung cancer (KrasG12V/+); Mammary cancer (Erbb2V664D, v-Hras), , ; Oligodendrogliomas (PDGF); B-cell lymphomas (Myc) |
| Cdk6 loss: | Lymphomas (v-Akt1) |
| Ccna2 loss: | Liver tumours (NRASG12V+53-shRNA) |
| Cdk2 loss: | Skin tumours (CDK4+DMBA+TPA); Mammary cancer (CCNE1LMW, Erbb2V664D), |
| Cdk1 loss: | Liver tumours (NRASG12V) |
| Chek1 loss: | Mammary cancer (Wnt1, Trp53+/−), ; Skin carcinomas (DMBA+TPA); Lymphomas (Chek2+/−); Skin papillomas (DMBA+TPA); Mammary cancer (Trp53+/−) |
| Chek2 loss: | B-cell lymphomas (Mus81−/−); Skin tumours (DMBA); Lung cancer; Mammary cancer (DMBA); Mammary cancer; Mammary cancer (Brca1−/−); T-cell lymphomas (Brca1−/−); Lymphomas (Chek1+/−) |
| Wee1 loss: | Mammary cancer |
| Plk1 loss: | Lymphomas |
| Plk3 loss: | Lung adenocarcinomas |
| Plk4 loss: | Hepatocellular carcinomas; Lung adenocarcinomas |
| Aurka loss: | Lymphomas |
| Aurkb loss: | Hepatocellular carcinomas, pituitary adenomas, skin papillomas |