

Sterile Neuroinflammation and Strategies for Therapeutic Intervention
- PMID: 28127491
- PMCID: PMC5239986
- DOI: 10.1155/2017/8385961
Sterile Neuroinflammation and Strategies for Therapeutic Intervention
Abstract
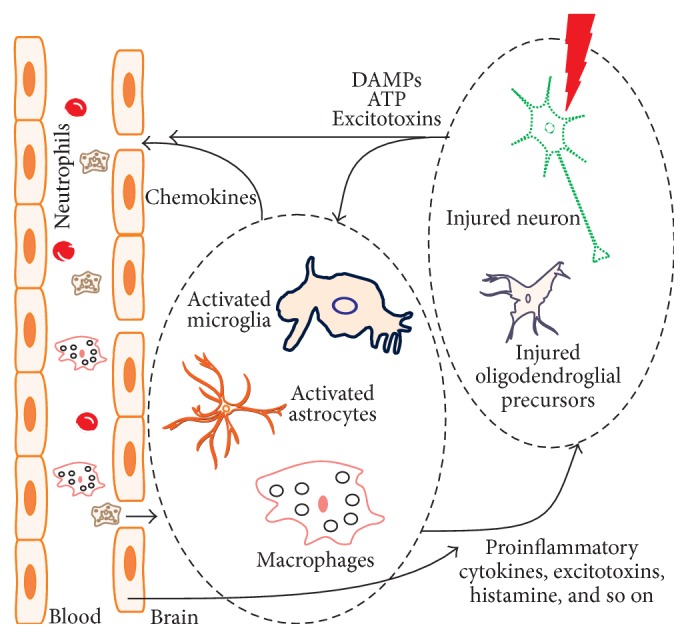
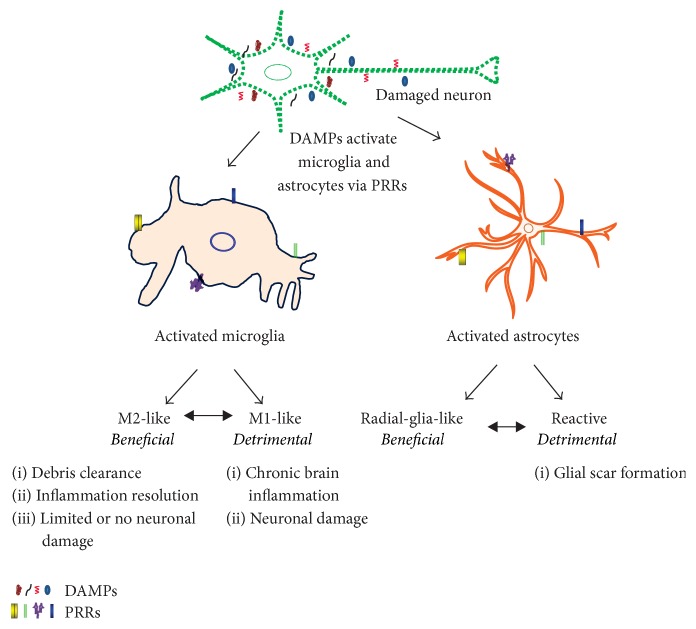
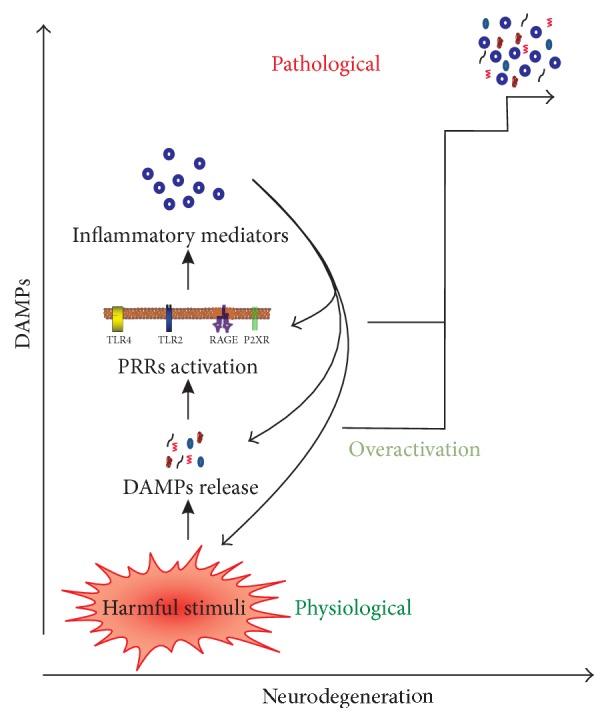
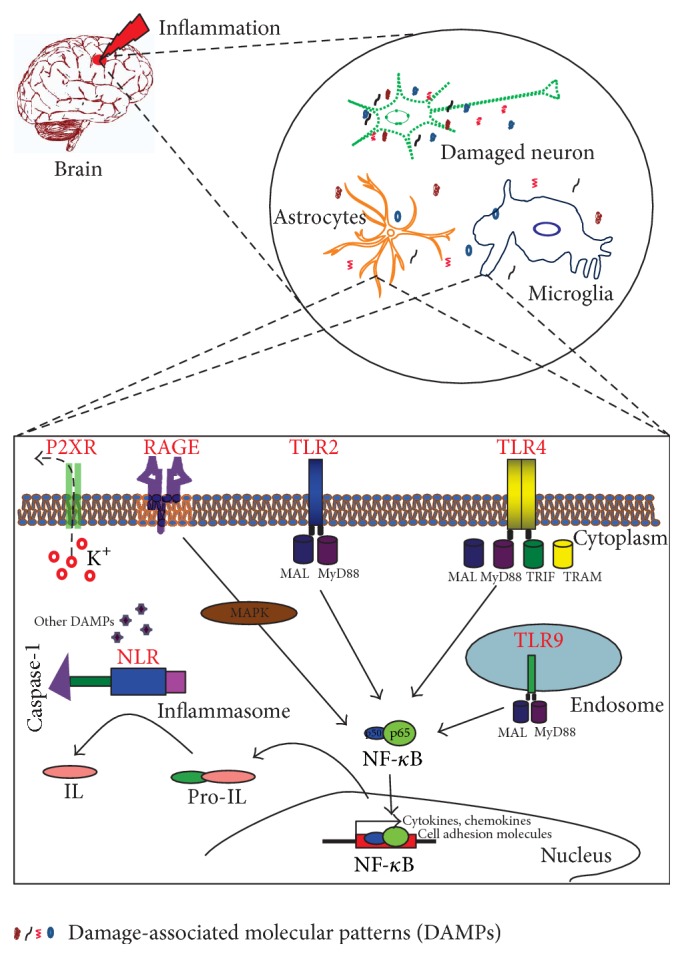
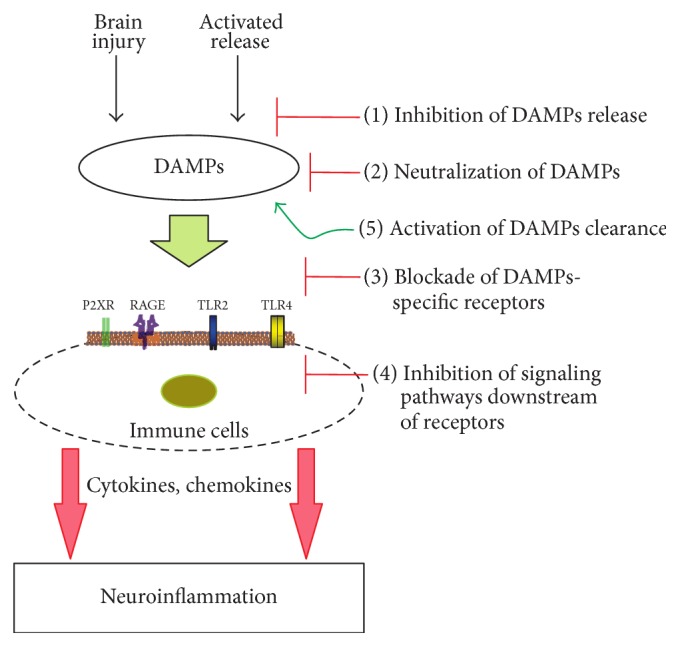
Sterile neuroinflammation is essential for the proper brain development and tissue repair. However, uncontrolled neuroinflammation plays a major role in the pathogenesis of various disease processes. The endogenous intracellular molecules so called damage-associated molecular patterns or alarmins or damage signals that are released by activated or necrotic cells are thought to play a crucial role in initiating an immune response. Sterile inflammatory response that occurs in Alzheimer's disease (AD), Parkinson's disease (PD), stroke, hemorrhage, epilepsy, or traumatic brain injury (TBI) creates a vicious cycle of unrestrained inflammation, driving progressive neurodegeneration. Neuroinflammation is a key mechanism in the progression (e.g., AD and PD) or secondary injury development (e.g., stroke, hemorrhage, stress, and TBI) of multiple brain conditions. Hence, it provides an opportunity for the therapeutic intervention to prevent progressive tissue damage and loss of function. The key for developing anti-neuroinflammatory treatment is to minimize the detrimental and neurotoxic effects of inflammation while promoting the beneficial and neurotropic effects, thereby creating ideal conditions for regeneration and repair. This review outlines how inflammation is involved in the pathogenesis of major nonpathogenic neuroinflammatory conditions and discusses the complex response of glial cells to damage signals. In addition, emerging experimental anti-neuroinflammatory drug treatment strategies are discussed.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Kitazawa M., Oddo S., Yamasaki T. R., Green K. N., LaFerla F. M. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer's disease. The Journal of Neuroscience. 2005;25(39):8843–8853. doi: 10.1523/jneurosci.2868-05.2005. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
