

Differential methylation of genes in individuals exposed to maternal diabetes in utero
- PMID: 28127622
- PMCID: PMC7194355
- DOI: 10.1007/s00125-016-4203-1
Differential methylation of genes in individuals exposed to maternal diabetes in utero
Abstract
Aims/hypothesis: Individuals exposed to maternal diabetes in utero are more likely to develop metabolic and cardiovascular diseases later in life. This may be partially attributable to epigenetic regulation of gene expression. We performed an epigenome-wide association study to examine whether differential DNA methylation, a major source of epigenetic regulation, can be observed in offspring of mothers with type 2 diabetes during the pregnancy (OMD) compared with offspring of mothers with no diabetes during the pregnancy (OMND).
Methods: DNA methylation was measured in peripheral blood using the Illumina HumanMethylation450K BeadChip. A total of 423,311 CpG sites were analysed in 388 Pima Indian individuals, mean age at examination was 13.0 years, 187 of whom were OMD and 201 were OMND. Differences in methylation between OMD and OMND were assessed.
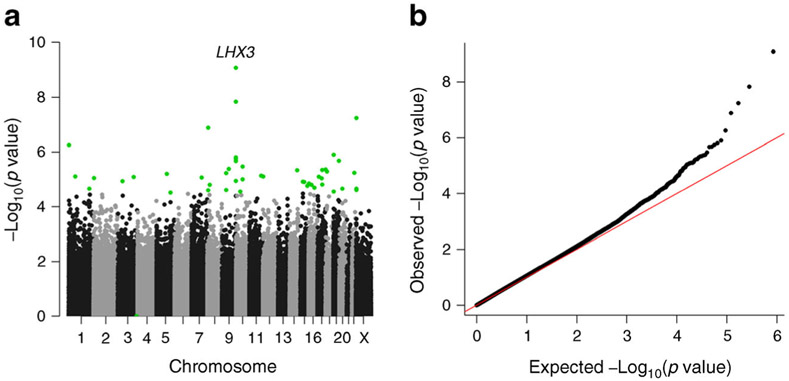
Results: Forty-eight differentially methylated CpG sites (with an empirical false discovery rate ≤0.05), mapping to 29 genes and ten intergenic regions, were identified. The gene with the strongest evidence was LHX3, in which six CpG sites were hypermethylated in OMD compared with OMND (p ≤ 1.1 × 10-5). Similarly, a CpG near PRDM16 was hypermethylated in OMD (1.1% higher, p = 5.6 × 10-7), where hypermethylation also predicted future diabetes risk (HR 2.12 per SD methylation increase, p = 9.7 × 10-5). Hypermethylation near AK3 and hypomethylation at PCDHGA4 and STC1 were associated with exposure to diabetes in utero (AK3: 2.5% higher, p = 7.8 × 10-6; PCDHGA4: 2.8% lower, p = 3.0 × 10-5; STC1: 2.9% lower, p = 1.6 × 10-5) and decreased insulin secretory function among offspring with normal glucose tolerance (AK3: 0.088 SD lower per SD of methylation increase, p = 0.02; PCDHGA4: 0.08 lower SD per SD of methylation decrease, p = 0.03; STC1: 0.072 SD lower per SD of methylation decrease, p = 0.05). Seventeen CpG sites were also associated with BMI (p ≤ 0.05). Pathway analysis of the genes with at least one differentially methylated CpG (p < 0.005) showed enrichment for three relevant biological pathways.
Conclusions/interpretation: Intrauterine exposure to diabetes can affect methylation at multiple genomic sites. Methylation status at some of these sites can impair insulin secretion, increase body weight and increase risk of type 2 diabetes.
Keywords: DNA methylation; Diabetes in pregnancy; Differentially methylated region; Maternal diabetes exposure in utero.
Figures
References
-
- Pettitt DJ, Aleck KA, Baird HR, Carraher MJ, Bennett PH, Knowler WC (1988) Congenital susceptibility to NIDDM. Role of intrauterine environment. Diabetes 37:622–628 - PubMed
-
- Dabelea D, Hanson RL, Lindsay RS et al. (2000) Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes 49:2208–2211 - PubMed
-
- Gautier JF, Wilson C, Weyer C et al. (2001) Low acute insulin secretory responses in adult offspring of people with early onset type 2 diabetes. Diabetes 50:1828–1833 - PubMed
-
- Ruchat SM, Hivert MF, Bouchard L (2013) Epigenetic programming of obesity and diabetes by in utero exposure to gestational diabetes mellitus. Nutr Rev 71(Suppl 1):S88–S94 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
