

CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo
- PMID: 28129107
- PMCID: PMC5363210
- DOI: 10.1016/j.ymthe.2016.11.010
CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo
Abstract
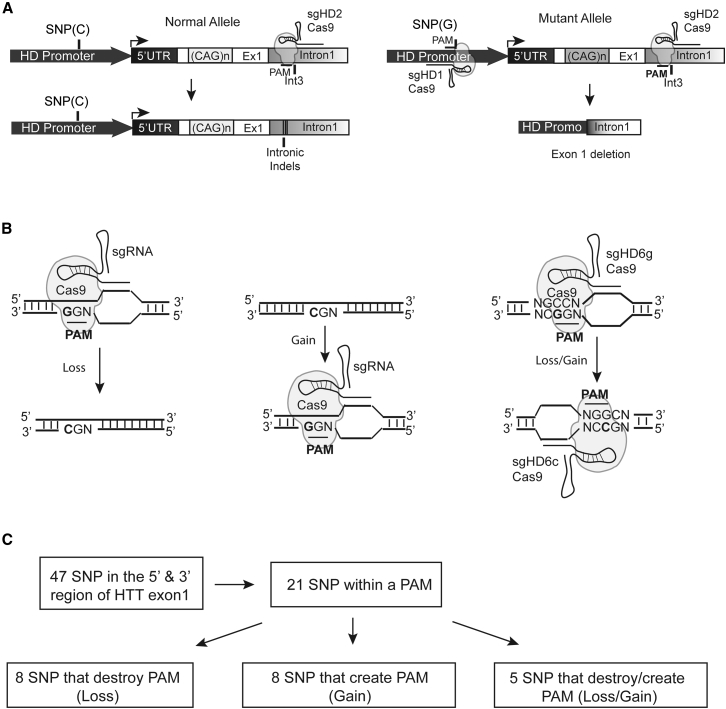
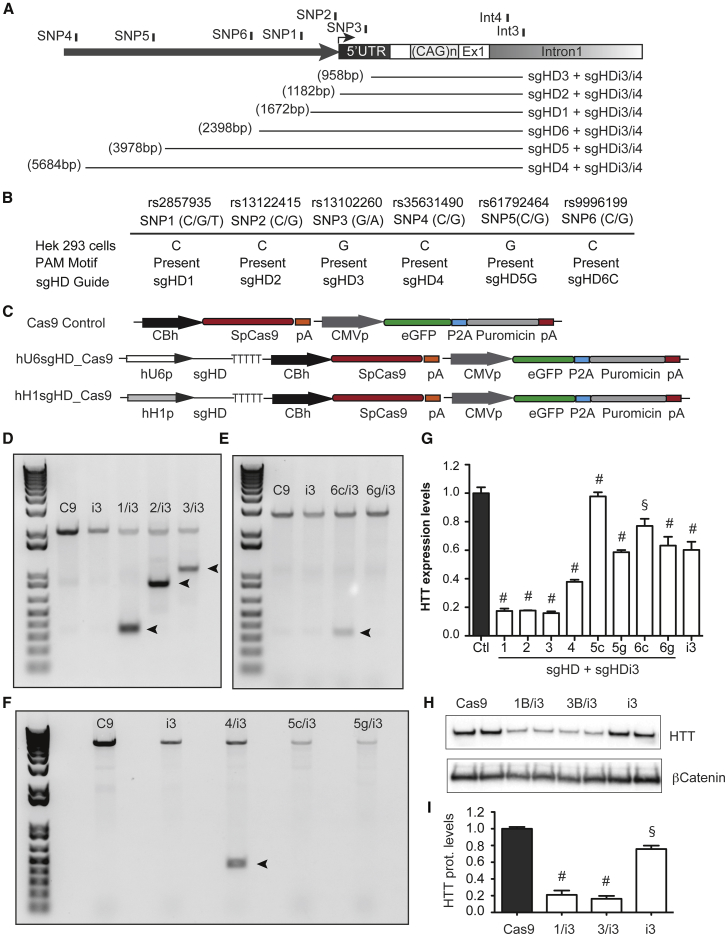
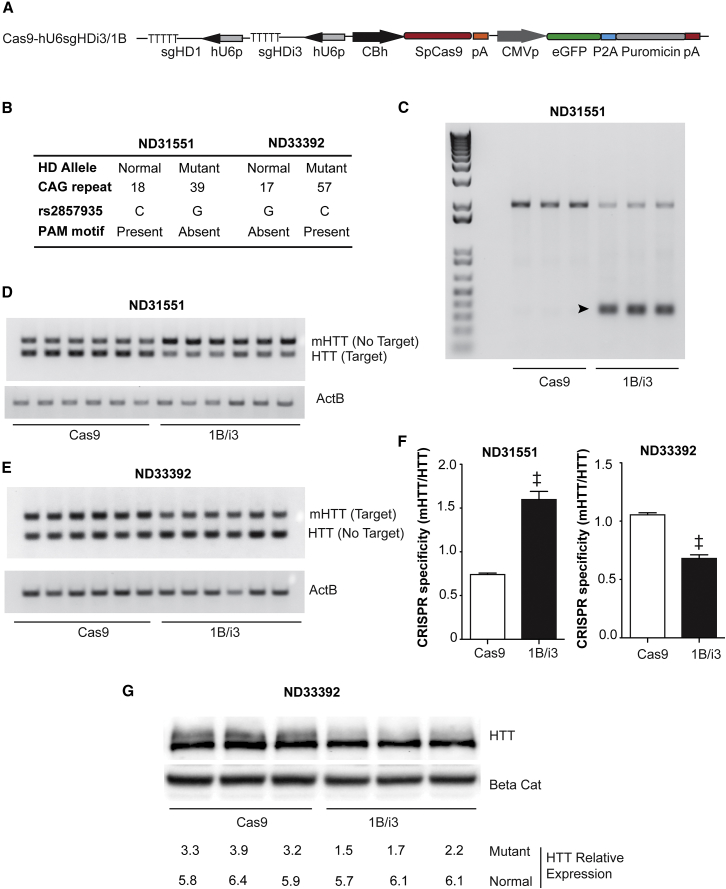
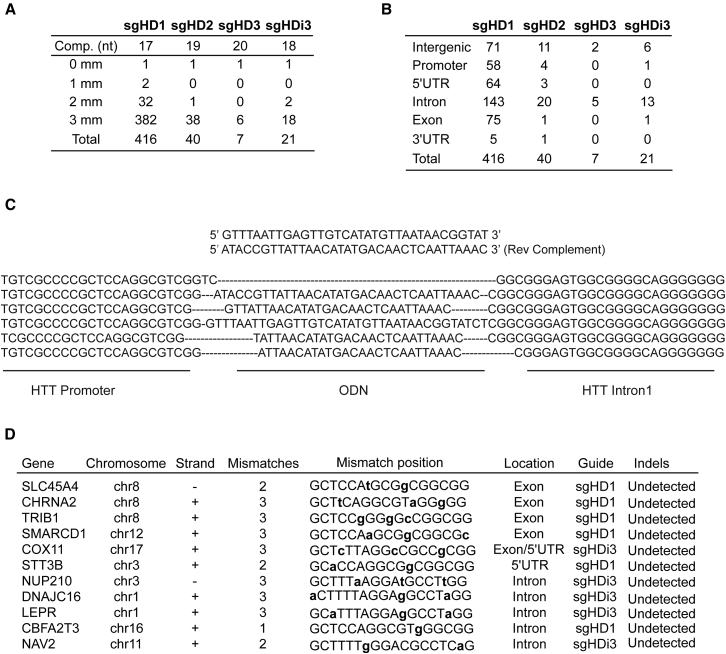
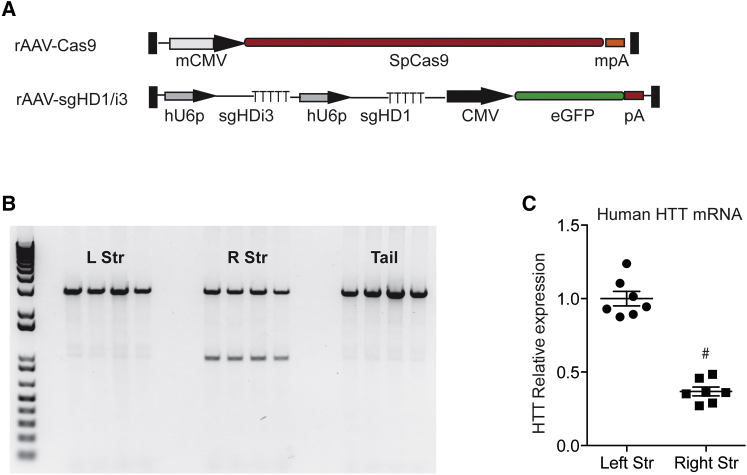
Huntington disease (HD) is a fatal dominantly inherited neurodegenerative disorder caused by CAG repeat expansion (>36 repeats) within the first exon of the huntingtin gene. Although mutant huntingtin (mHTT) is ubiquitously expressed, the brain shows robust and early degeneration. Current RNA interference-based approaches for lowering mHTT expression have been efficacious in mouse models, but basal mutant protein levels are still detected. To fully mitigate expression from the mutant allele, we hypothesize that allele-specific genome editing can occur via prevalent promoter-resident SNPs in heterozygosity with the mutant allele. Here, we identified SNPs that either cause or destroy PAM motifs critical for CRISPR-selective editing of one allele versus the other in cells from HD patients and in a transgenic HD model harboring the human allele.
Keywords: AAV; CRISPR/Cas9; Huntington’s disease; gene therapy; genome editing.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Walker F.O. Huntington’s disease. Semin. Neurol. 2007;27:143–150. - PubMed
-
- Hicks R.R., Smith D.H., Lowenstein D.H., Saint Marie R., McIntosh T.K. Mild experimental brain injury in the rat induces cognitive deficits associated with regional neuronal loss in the hippocampus. J. Neurotrauma. 1993;10:405–414. - PubMed
-
- Yamamoto A., Lucas J.J., Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell. 2000;101:57–66. - PubMed
-
- Díaz-Hernández M., Torres-Peraza J., Salvatori-Abarca A., Morán M.A., Gómez-Ramos P., Alberch J., Lucas J.J. Full motor recovery despite striatal neuron loss and formation of irreversible amyloid-like inclusions in a conditional mouse model of Huntington’s disease. J. Neurosci. 2005;25:9773–9781. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
