

Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis
- PMID: 28132687
- PMCID: PMC5294679
- DOI: 10.1016/j.ajhg.2017.01.005
Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis
Abstract
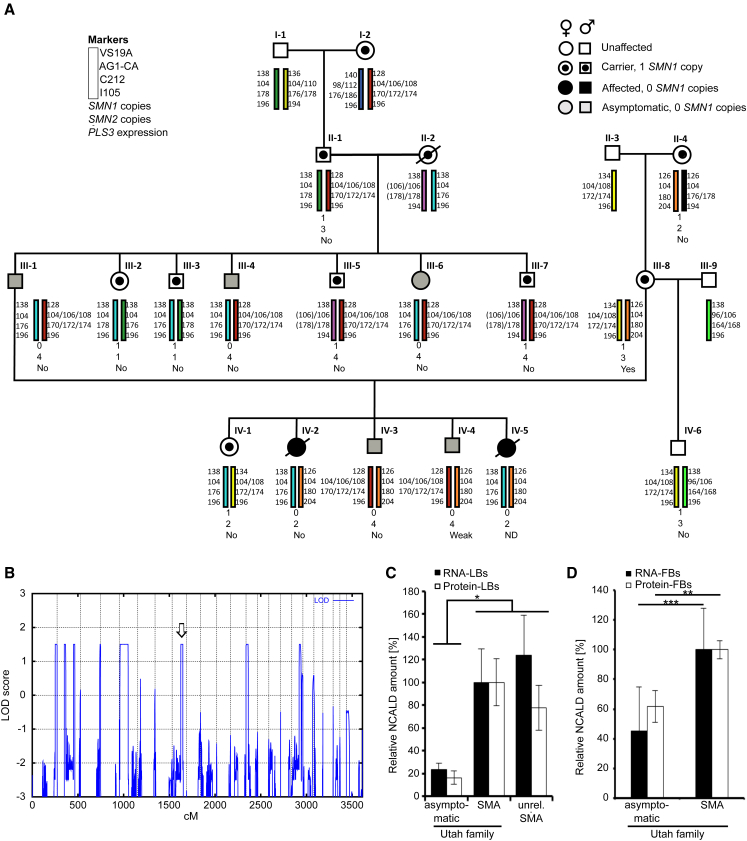
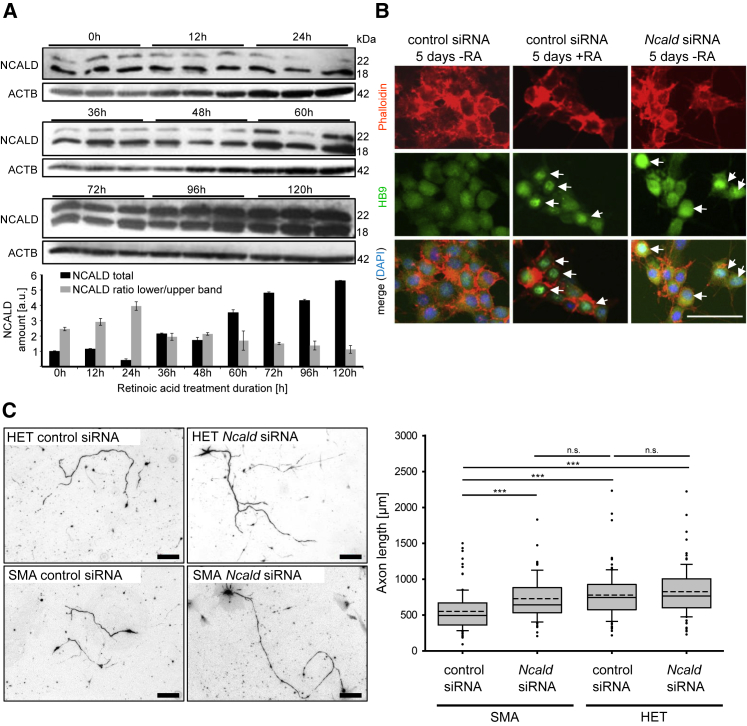
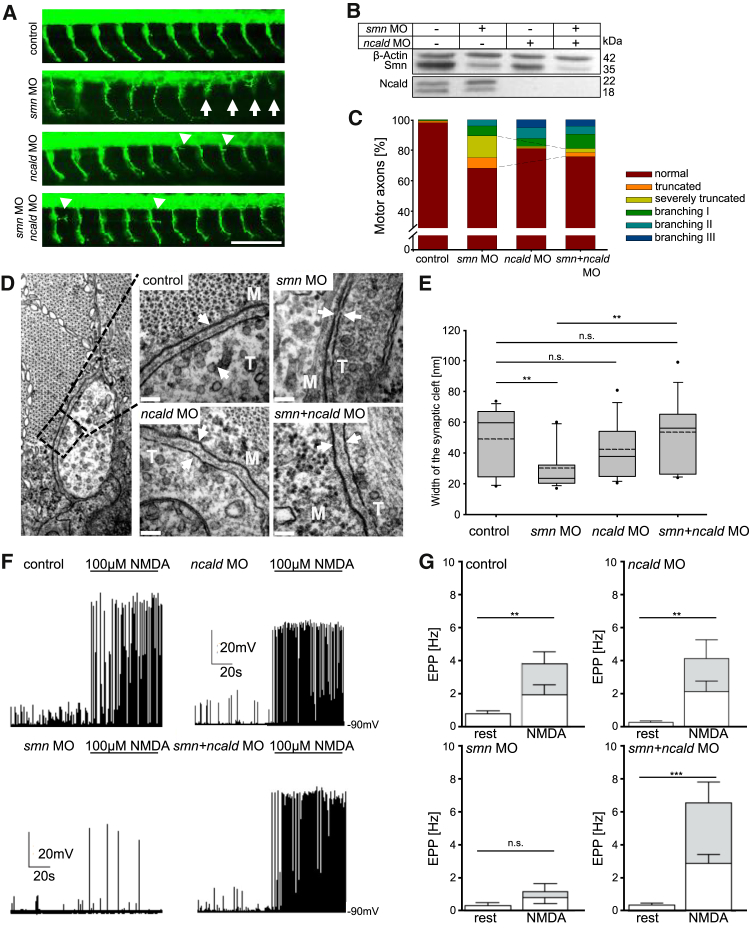
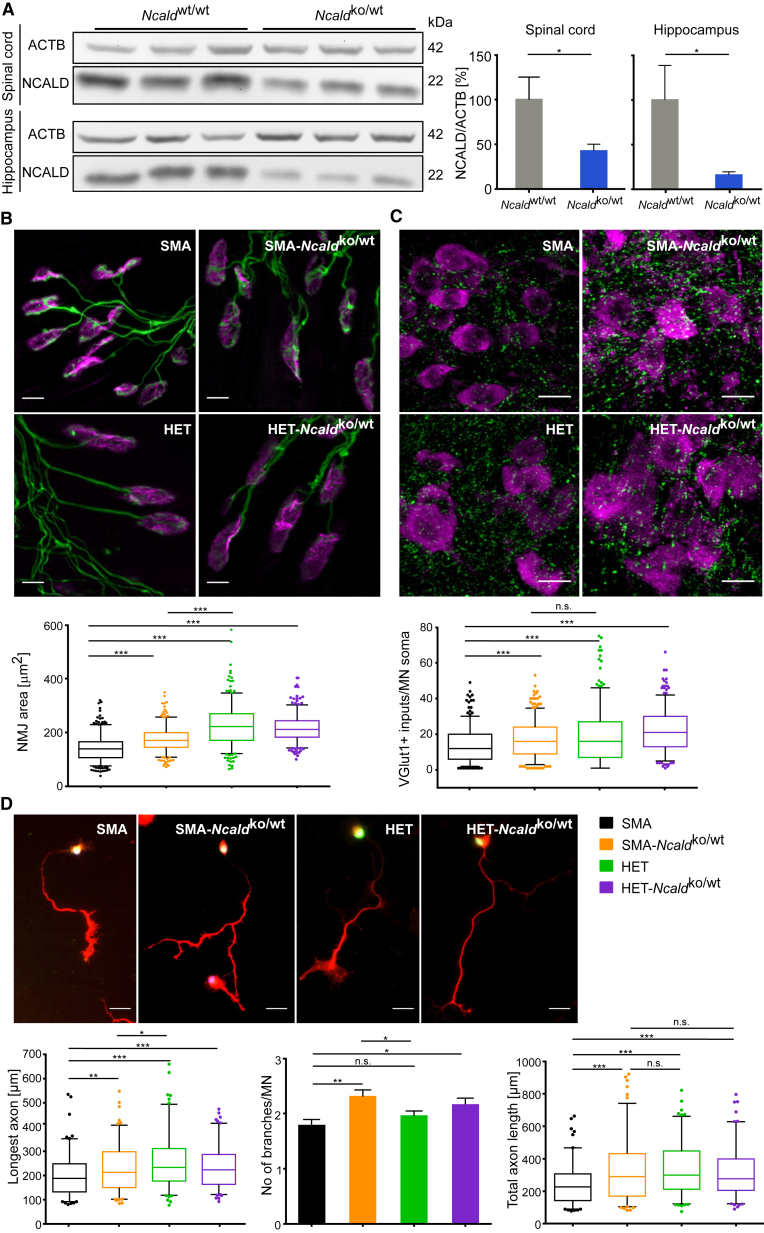
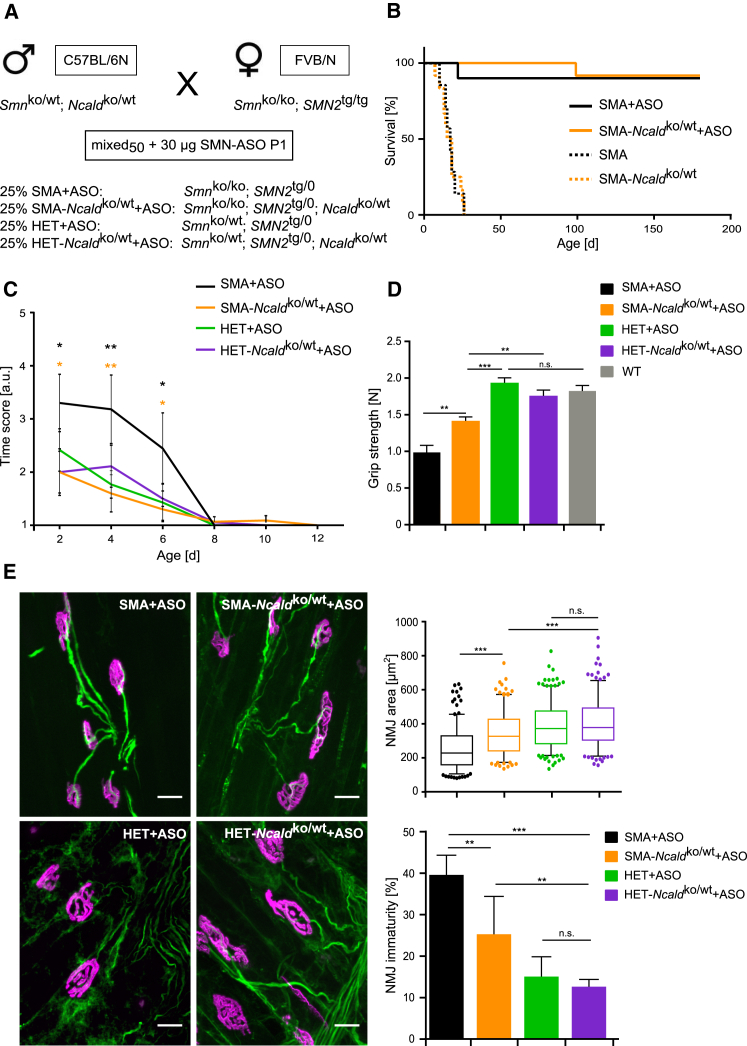
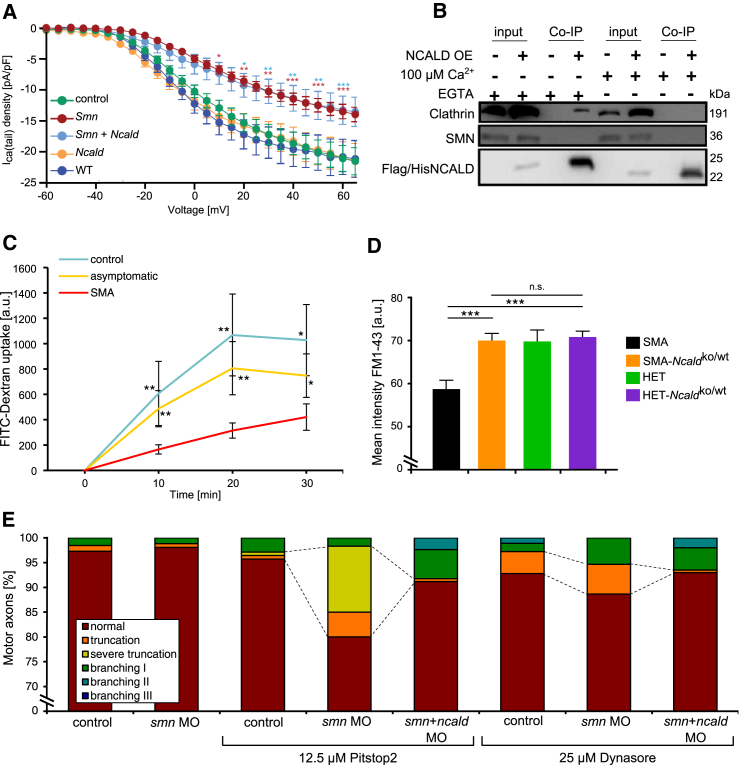
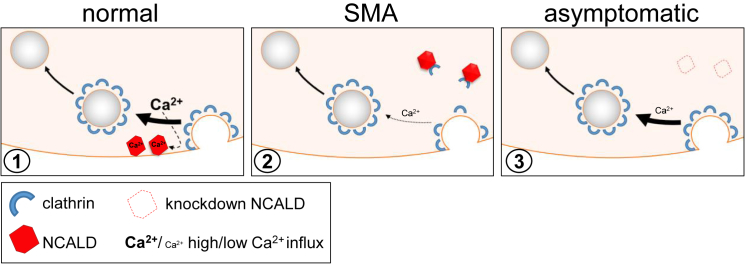
Homozygous SMN1 loss causes spinal muscular atrophy (SMA), the most common lethal genetic childhood motor neuron disease. SMN1 encodes SMN, a ubiquitous housekeeping protein, which makes the primarily motor neuron-specific phenotype rather unexpected. SMA-affected individuals harbor low SMN expression from one to six SMN2 copies, which is insufficient to functionally compensate for SMN1 loss. However, rarely individuals with homozygous absence of SMN1 and only three to four SMN2 copies are fully asymptomatic, suggesting protection through genetic modifier(s). Previously, we identified plastin 3 (PLS3) overexpression as an SMA protective modifier in humans and showed that SMN deficit impairs endocytosis, which is rescued by elevated PLS3 levels. Here, we identify reduction of the neuronal calcium sensor Neurocalcin delta (NCALD) as a protective SMA modifier in five asymptomatic SMN1-deleted individuals carrying only four SMN2 copies. We demonstrate that NCALD is a Ca2+-dependent negative regulator of endocytosis, as NCALD knockdown improves endocytosis in SMA models and ameliorates pharmacologically induced endocytosis defects in zebrafish. Importantly, NCALD knockdown effectively ameliorates SMA-associated pathological defects across species, including worm, zebrafish, and mouse. In conclusion, our study identifies a previously unknown protective SMA modifier in humans, demonstrates modifier impact in three different SMA animal models, and suggests a potential combinatorial therapeutic strategy to efficiently treat SMA. Since both protective modifiers restore endocytosis, our results confirm that endocytosis is a major cellular mechanism perturbed in SMA and emphasize the power of protective modifiers for understanding disease mechanism and developing therapies.
Keywords: NCALD; PLS3; SMA; SMN1; SMN2; asymptomatic; endocytosis; genetic modifier; incomplete penetrance; neuronal sensor protein; spinal muscular dystrophy.
Copyright © 2017 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Wirth B., Garbes L., Riessland M. How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Curr. Opin. Genet. Dev. 2013;23:330–338. - PubMed
-
- Kaczmarek A., Schneider S., Wirth B., Riessland M. Investigational therapies for the treatment of spinal muscular atrophy. Expert Opin. Investig. Drugs. 2015;24:867–881. - PubMed
-
- Lefebvre S., Bürglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. - PubMed
-
- Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA) Hum. Mutat. 2000;15:228–237. - PubMed
-
- Liu Q., Fischer U., Wang F., Dreyfuss G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell. 1997;90:1013–1021. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
