

Active Interaction Mapping Reveals the Hierarchical Organization of Autophagy
- PMID: 28132844
- PMCID: PMC5439305
- DOI: 10.1016/j.molcel.2016.12.024
Active Interaction Mapping Reveals the Hierarchical Organization of Autophagy
Abstract
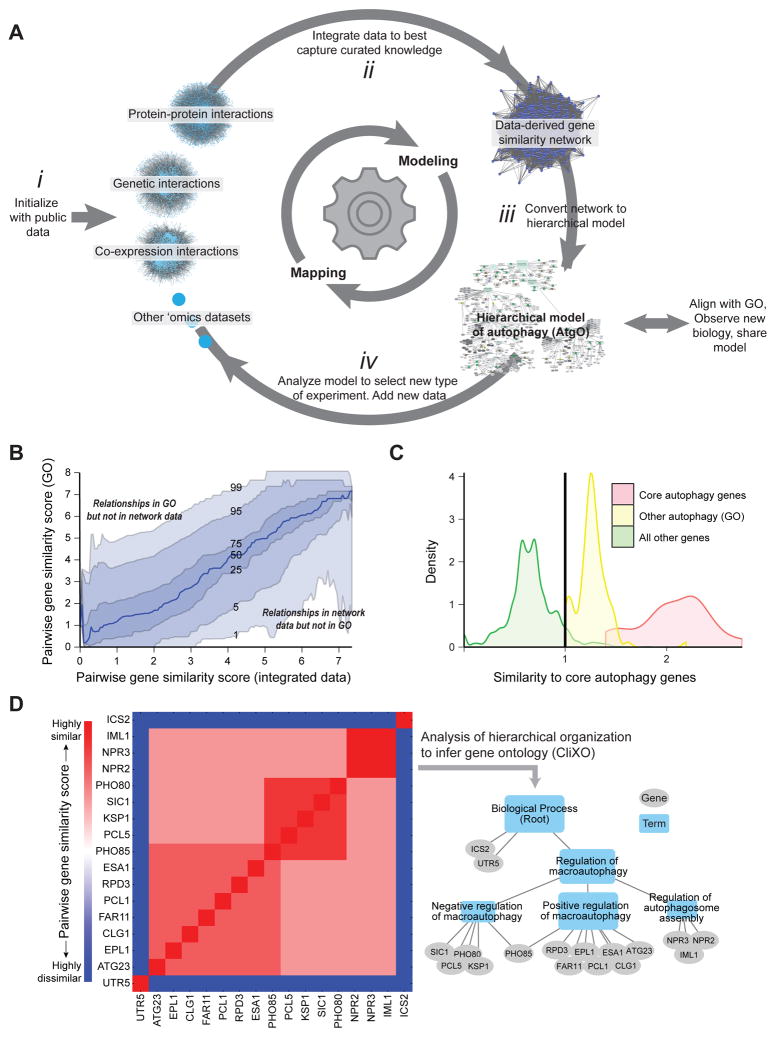
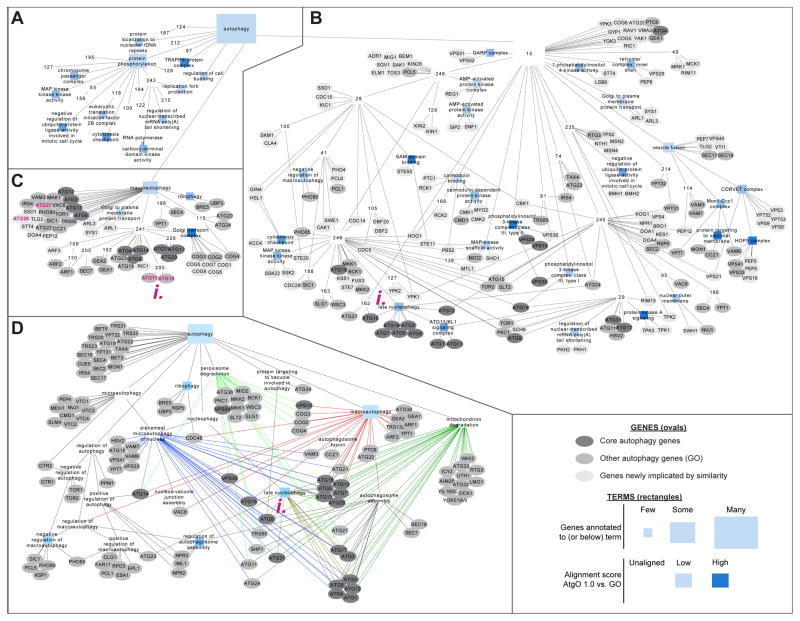
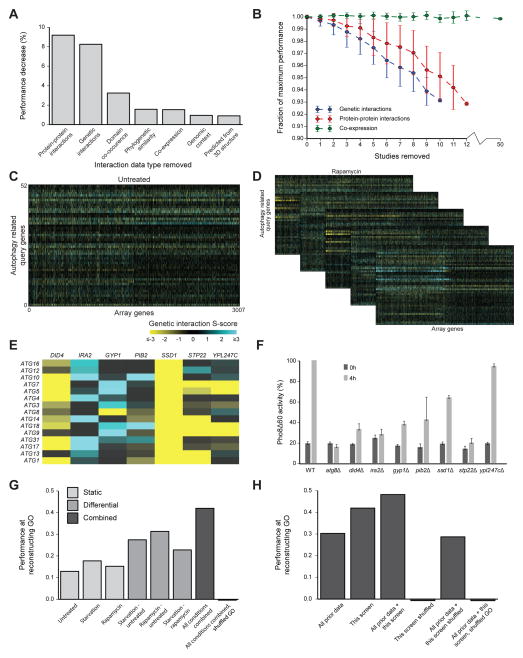
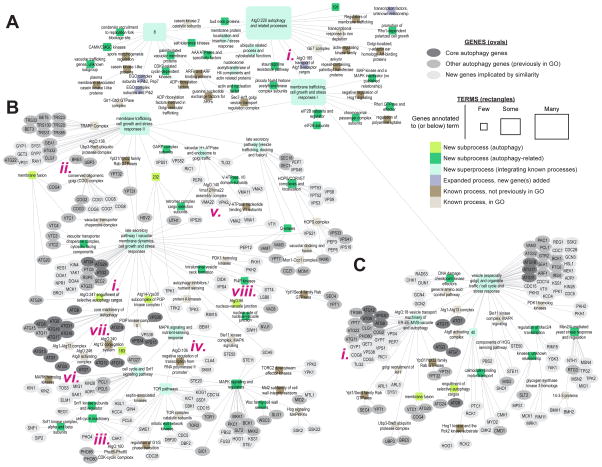
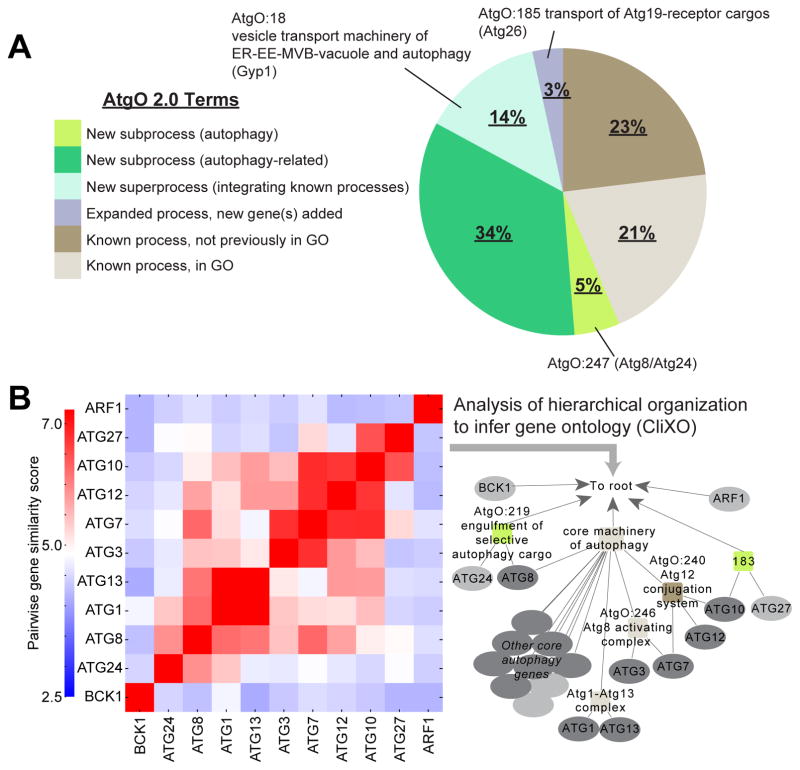
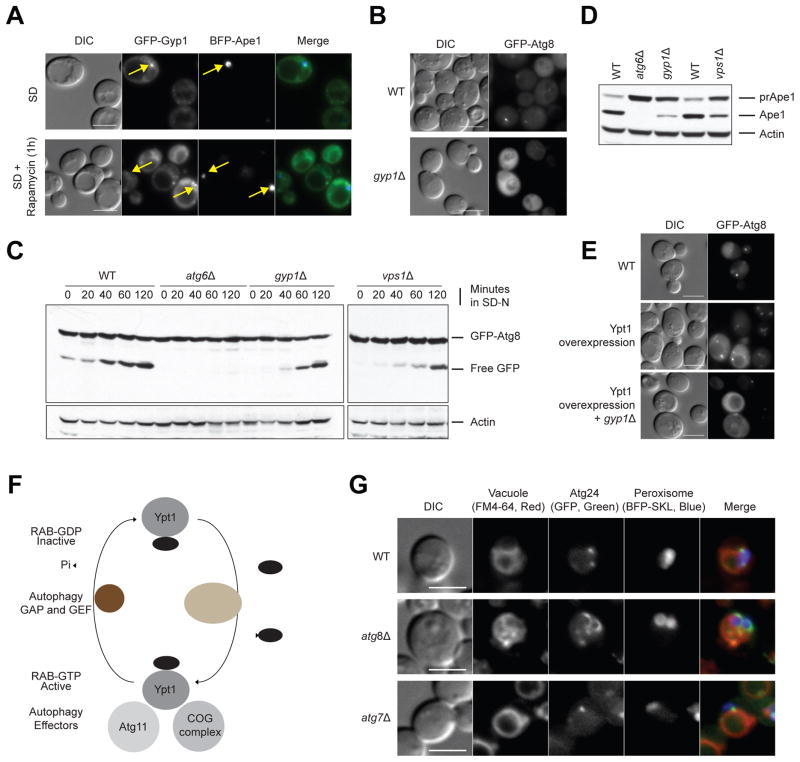
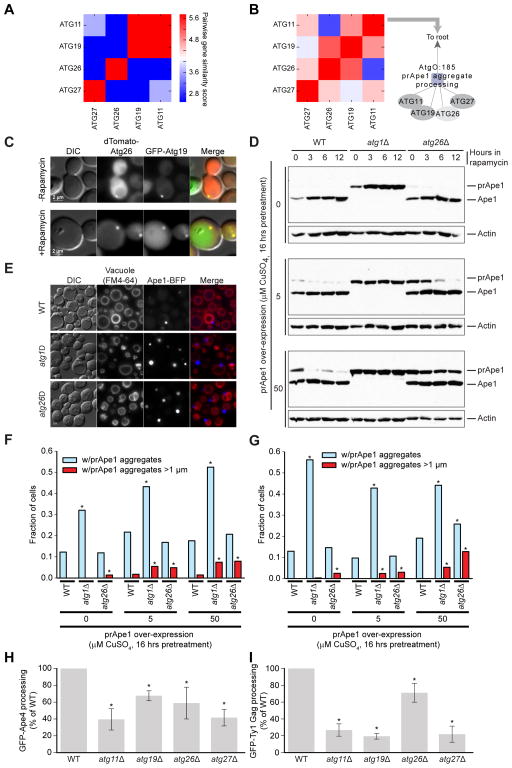
We have developed a general progressive procedure, Active Interaction Mapping, to guide assembly of the hierarchy of functions encoding any biological system. Using this process, we assemble an ontology of functions comprising autophagy, a central recycling process implicated in numerous diseases. A first-generation model, built from existing gene networks in Saccharomyces, captures most known autophagy components in broad relation to vesicle transport, cell cycle, and stress response. Systematic analysis identifies synthetic-lethal interactions as most informative for further experiments; consequently, we saturate the model with 156,364 such measurements across autophagy-activating conditions. These targeted interactions provide more information about autophagy than all previous datasets, producing a second-generation ontology of 220 functions. Approximately half are previously unknown; we confirm roles for Gyp1 at the phagophore-assembly site, Atg24 in cargo engulfment, Atg26 in cytoplasm-to-vacuole targeting, and Ssd1, Did4, and others in selective and non-selective autophagy. The procedure and autophagy hierarchy are at http://atgo.ucsd.edu/.
Keywords: active interaction mapping; autophagy; hierarchical modeling; human; systems biology; yeast.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
