

DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma
- PMID: 28134926
- PMCID: PMC5951283
- DOI: 10.1038/nm.4273
DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma
Abstract
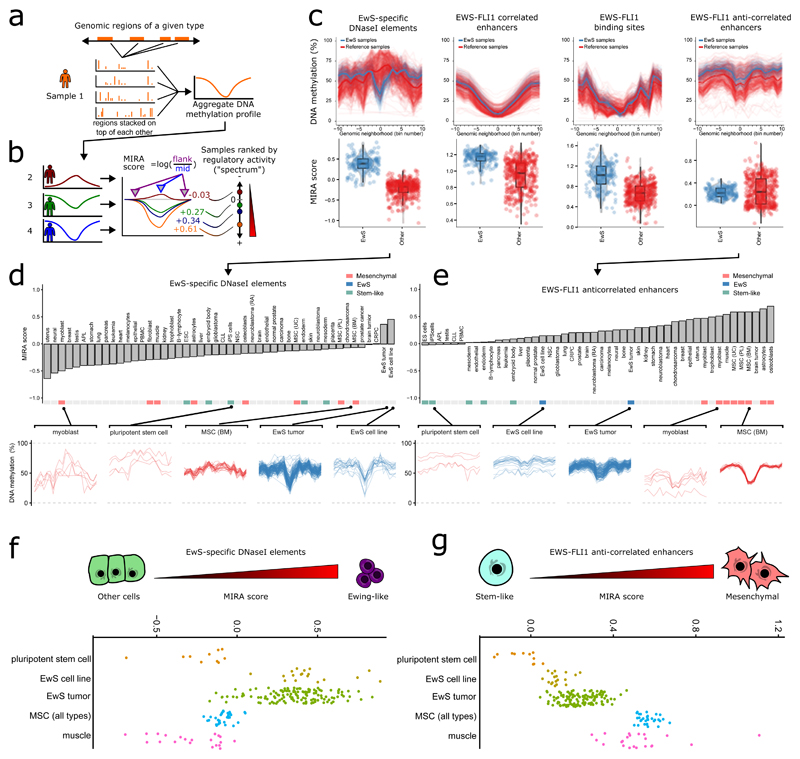
Developmental tumors in children and young adults carry few genetic alterations, yet they have diverse clinical presentation. Focusing on Ewing sarcoma, we sought to establish the prevalence and characteristics of epigenetic heterogeneity in genetically homogeneous cancers. We performed genome-scale DNA methylation sequencing for a large cohort of Ewing sarcoma tumors and analyzed epigenetic heterogeneity on three levels: between cancers, between tumors, and within tumors. We observed consistent DNA hypomethylation at enhancers regulated by the disease-defining EWS-FLI1 fusion protein, thus establishing epigenomic enhancer reprogramming as a ubiquitous and characteristic feature of Ewing sarcoma. DNA methylation differences between tumors identified a continuous disease spectrum underlying Ewing sarcoma, which reflected the strength of an EWS-FLI1 regulatory signature and a continuum between mesenchymal and stem cell signatures. There was substantial epigenetic heterogeneity within tumors, particularly in patients with metastatic disease. In summary, our study provides a comprehensive assessment of epigenetic heterogeneity in Ewing sarcoma and thereby highlights the importance of considering nongenetic aspects of tumor heterogeneity in the context of cancer biology and personalized medicine.
Conflict of interest statement
The authors declare no competing financial interests.
Figures




References
-
- de Álava E, Lessnick SL, Sorensen PHB. Ewing sarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. WHO Classification of tumours of soft tissue and bone. 2013. pp. 306–309.
-
- Schmidt D, Herrmann C, Jürgens H, Harms D. Malignant peripheral neuroectodermal tumor and its necessary distinction from Ewing’s sarcoma. A report from the Kiel Pediatric Tumor Registry. Cancer. 1991;68:2251–2259. doi: 10.1002/1097-0142(19911115)68:10%3c2251::AID-CNCR2820681025%3e3.0.CO;2-X. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
