

Ischemia/Reperfusion
- PMID: 28135002
- PMCID: PMC5648017
- DOI: 10.1002/cphy.c160006
Ischemia/Reperfusion
Abstract
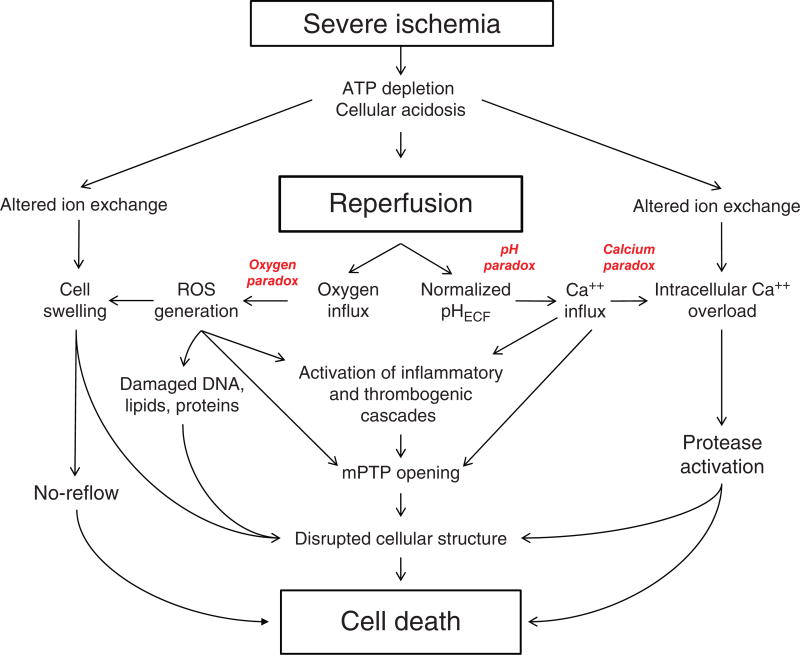
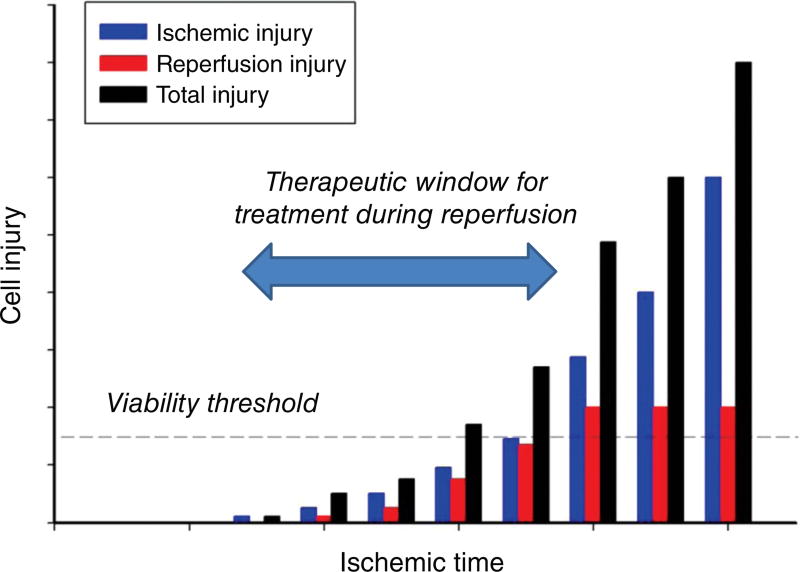
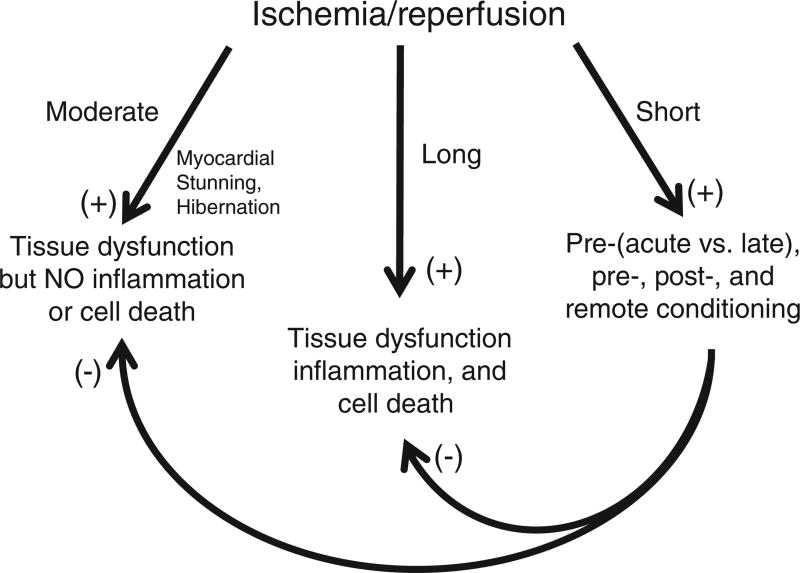
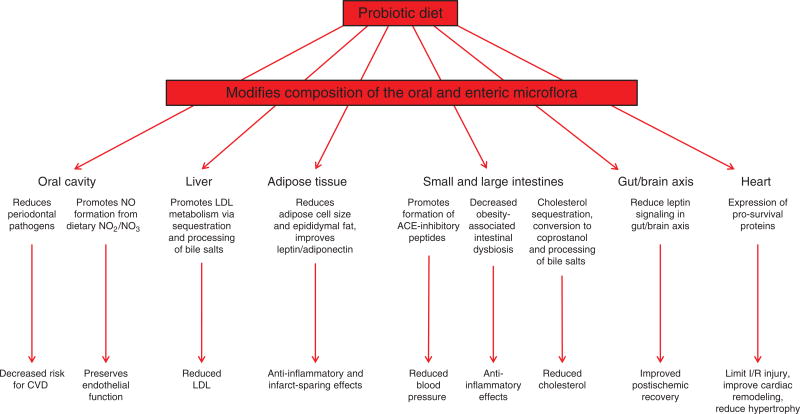
Ischemic disorders, such as myocardial infarction, stroke, and peripheral vascular disease, are the most common causes of debilitating disease and death in westernized cultures. The extent of tissue injury relates directly to the extent of blood flow reduction and to the length of the ischemic period, which influence the levels to which cellular ATP and intracellular pH are reduced. By impairing ATPase-dependent ion transport, ischemia causes intracellular and mitochondrial calcium levels to increase (calcium overload). Cell volume regulatory mechanisms are also disrupted by the lack of ATP, which can induce lysis of organelle and plasma membranes. Reperfusion, although required to salvage oxygen-starved tissues, produces paradoxical tissue responses that fuel the production of reactive oxygen species (oxygen paradox), sequestration of proinflammatory immunocytes in ischemic tissues, endoplasmic reticulum stress, and development of postischemic capillary no-reflow, which amplify tissue injury. These pathologic events culminate in opening of mitochondrial permeability transition pores as a common end-effector of ischemia/reperfusion (I/R)-induced cell lysis and death. Emerging concepts include the influence of the intestinal microbiome, fetal programming, epigenetic changes, and microparticles in the pathogenesis of I/R. The overall goal of this review is to describe these and other mechanisms that contribute to I/R injury. Because so many different deleterious events participate in I/R, it is clear that therapeutic approaches will be effective only when multiple pathologic processes are targeted. In addition, the translational significance of I/R research will be enhanced by much wider use of animal models that incorporate the complicating effects of risk factors for cardiovascular disease. © 2017 American Physiological Society. Compr Physiol 7:113-170, 2017.
Copyright © 2017 John Wiley & Sons, Inc.
Figures
References
-
- Abela CB, Homer-Vanniasinkham S. Clinical implications of ischaemia-reperfusion injury. Pathophysiology. 2003;9:229–240. - PubMed
-
- Abonia JP, Friend DS, Austen WG, Jr, Moore FD, Jr, Carroll MC, Chan R, Afnan J, Humbles A, Gerard C, Knight P, Kanaoka Y, Yasuda S, Morokawa N, Austen KF, Stevens RL, Gurish MF. Mast cell protease 5 mediates ischemia-reperfusion injury of mouse skeletal muscle. J Immunol. 2005;174:7285–7291. - PMC - PubMed
-
- Adibhatha RM, Hatche JF. Lipid oxidation and peroxidation in CNS health and disease: From molecular mechanisms to therapeutic opportunities. Antiox Redox Signal. 2010;12:125–169. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
