

Genotyping-by-Sequencing in a Species Complex of Australian Hummock Grasses (Triodia): Methodological Insights and Phylogenetic Resolution
- PMID: 28135342
- PMCID: PMC5279811
- DOI: 10.1371/journal.pone.0171053
Genotyping-by-Sequencing in a Species Complex of Australian Hummock Grasses (Triodia): Methodological Insights and Phylogenetic Resolution
Abstract
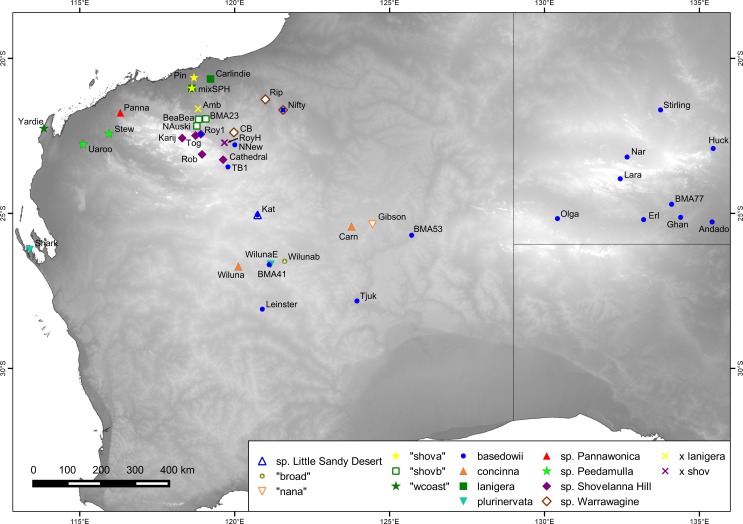
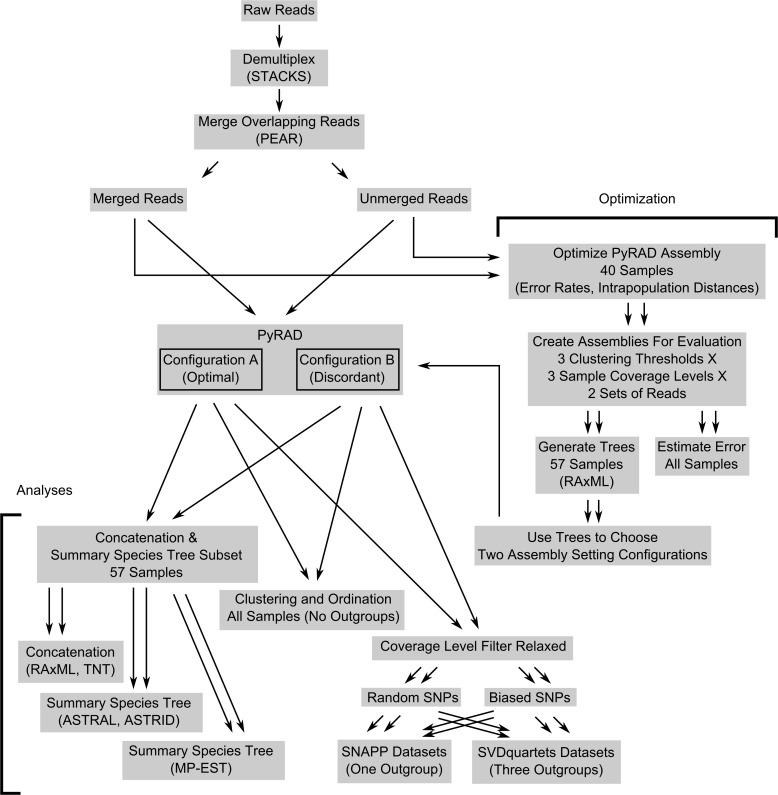
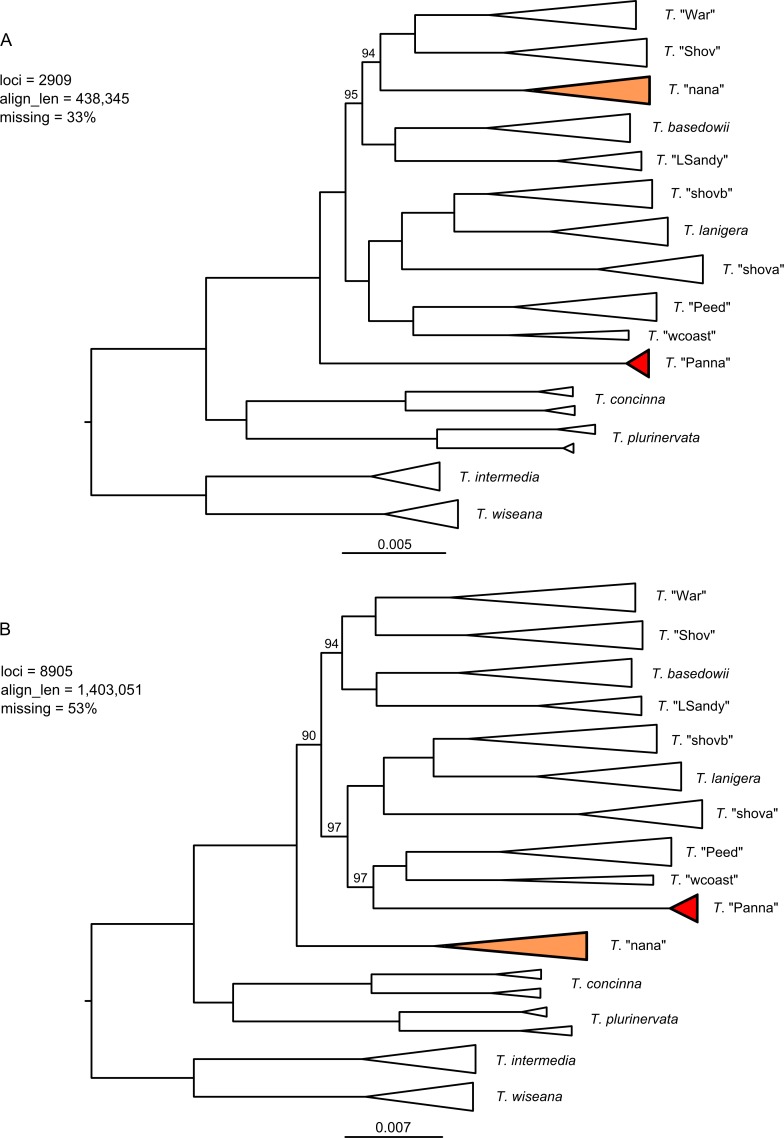
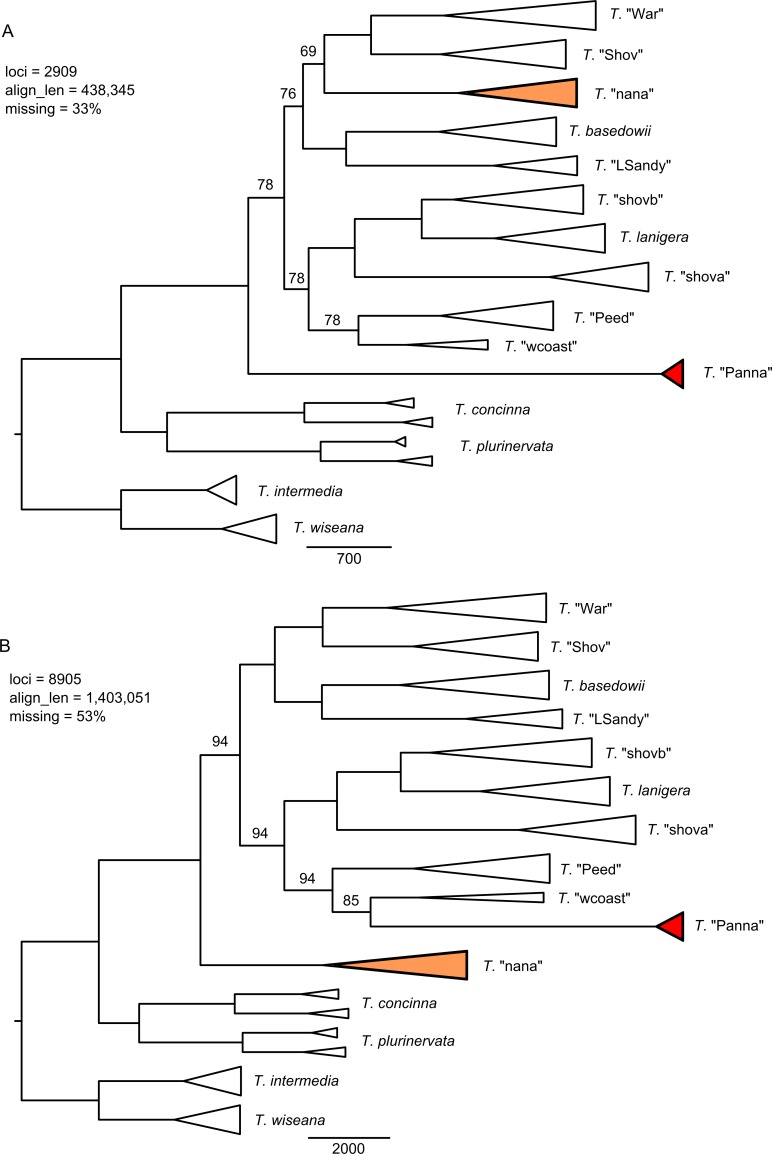
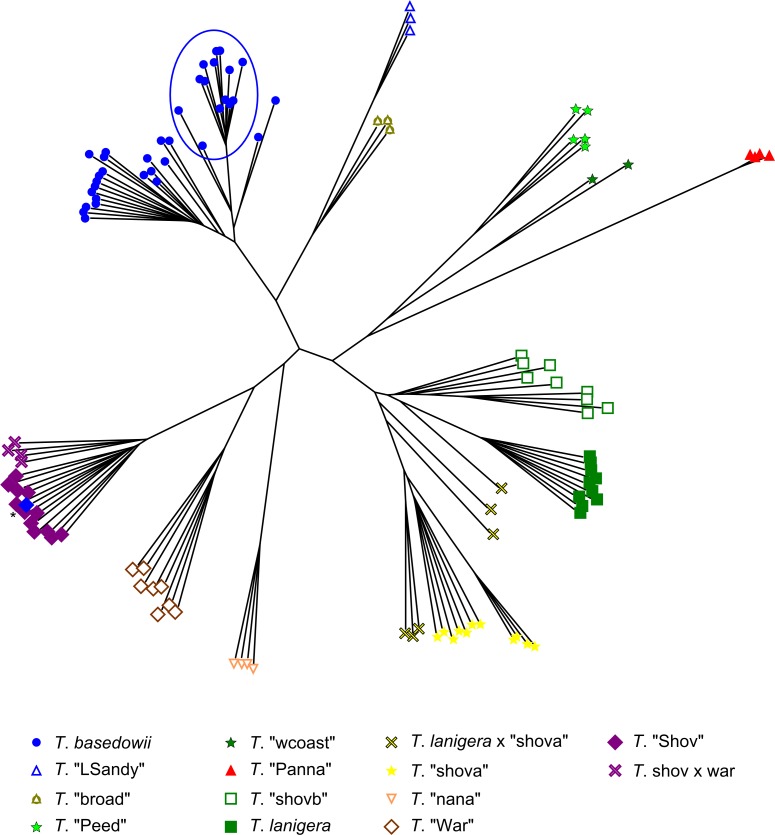
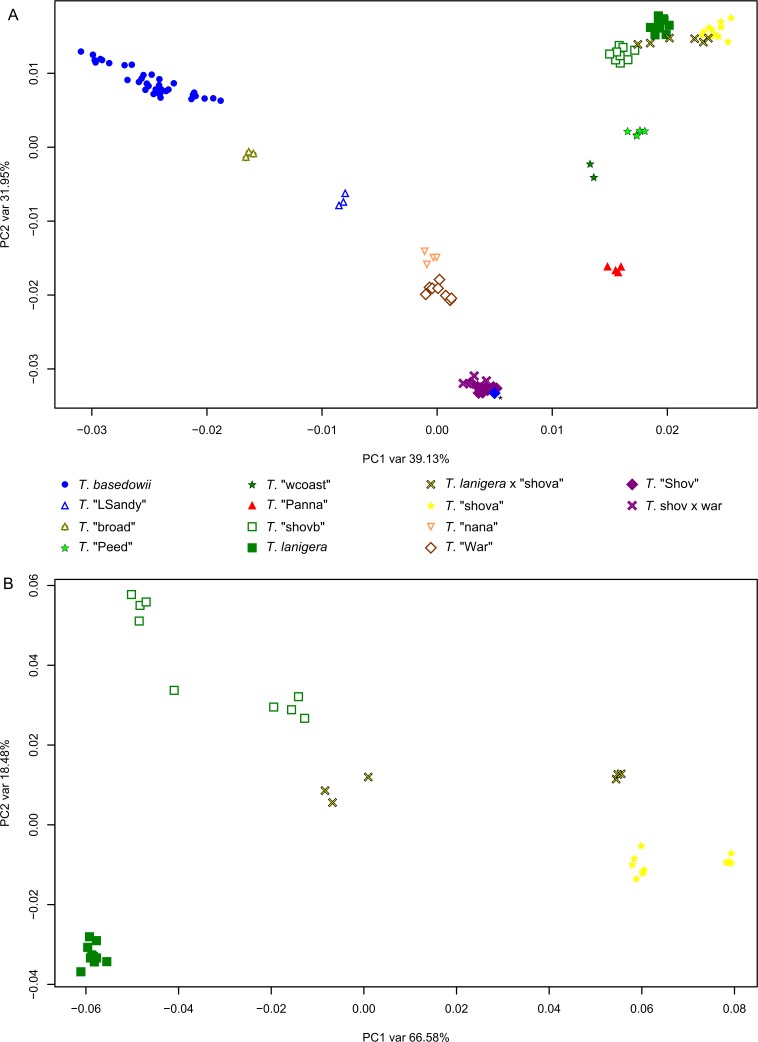
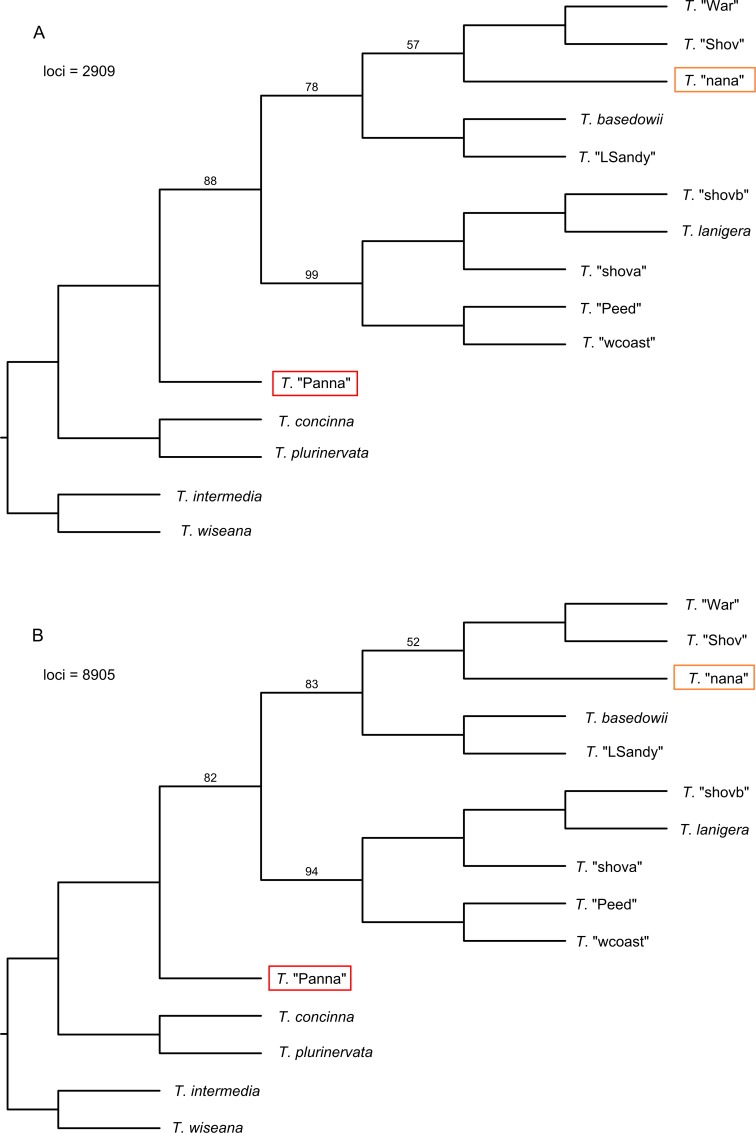
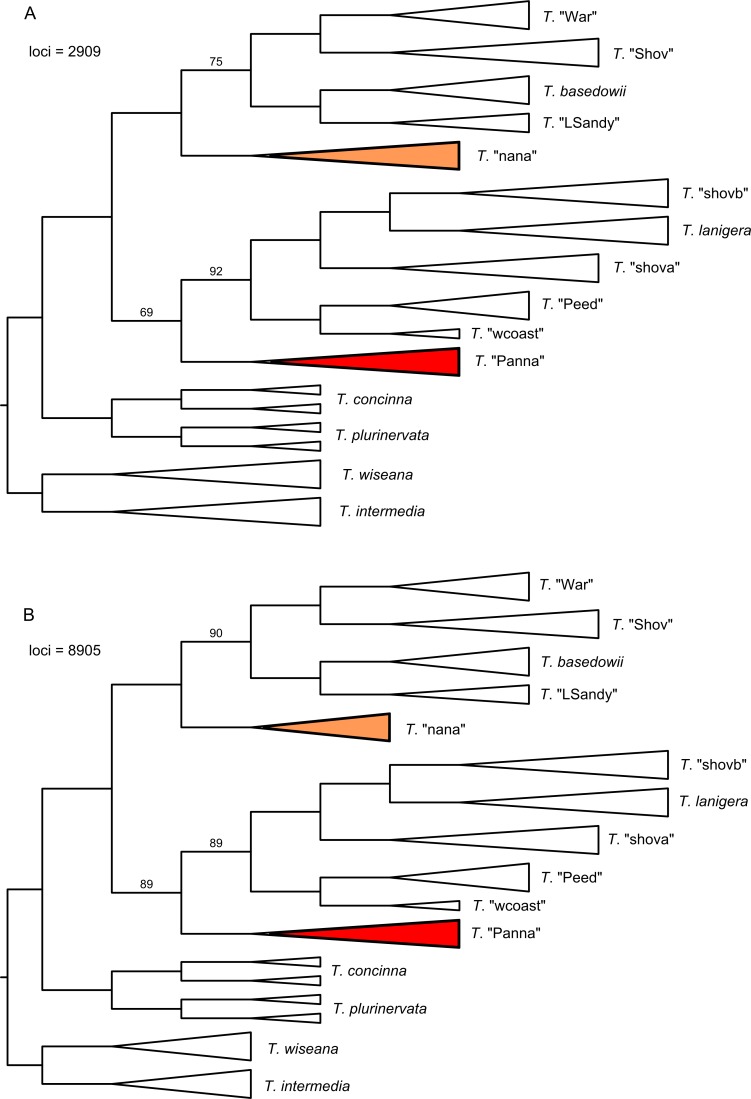
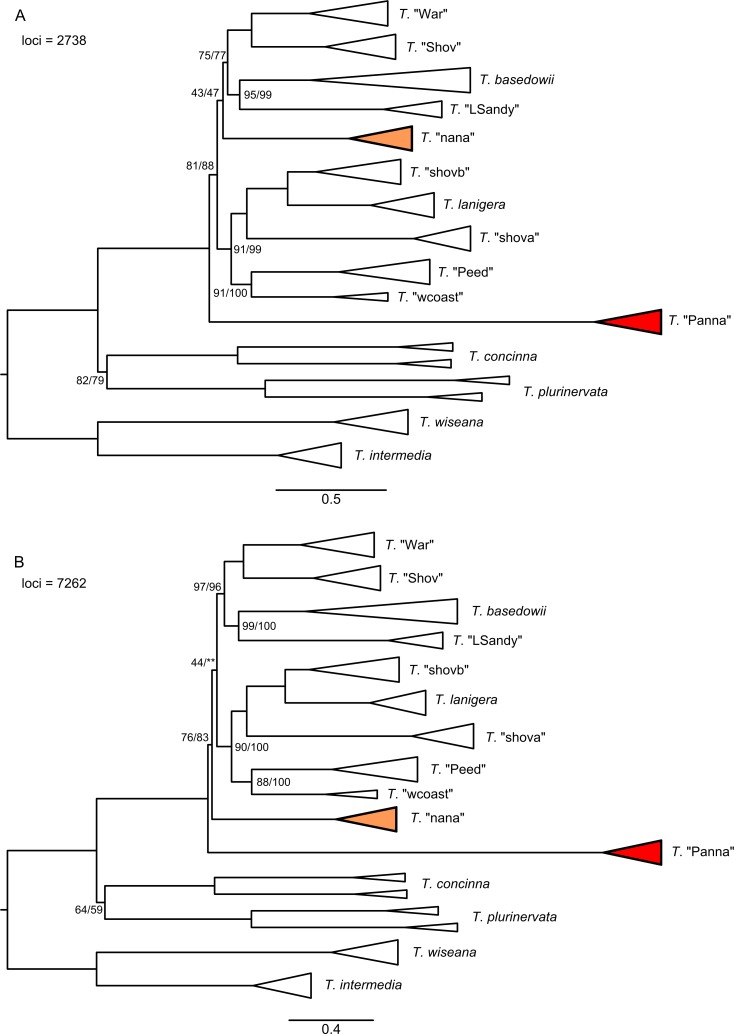
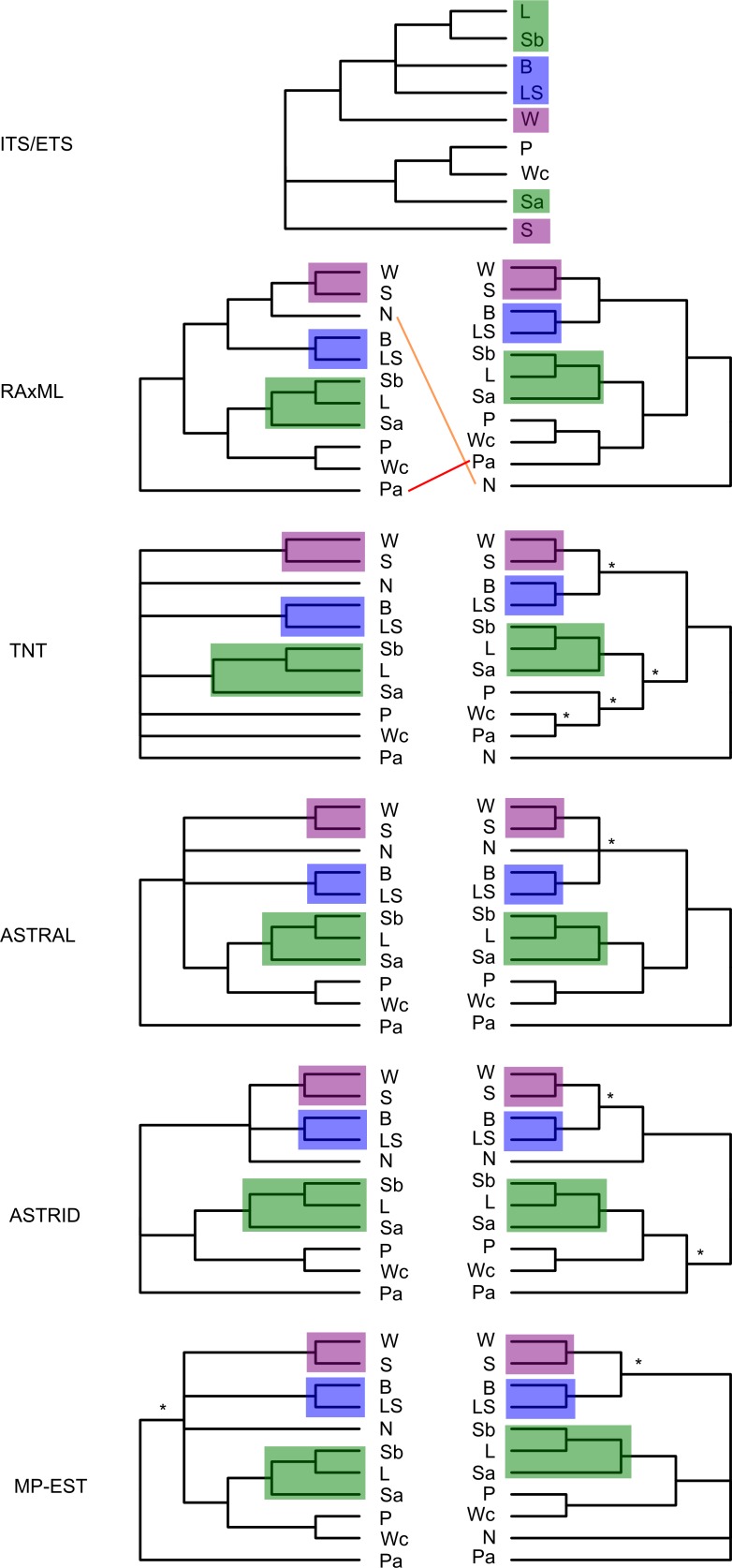
Next-generation sequencing is becoming increasingly accessible to researchers asking biosystematic questions, but current best practice in both choosing a specific approach and effectively analysing the resulting data set is still being explored. We present a case study for the use of genotyping-by-sequencing (GBS) to resolve relationships in a species complex of Australian arid and semi-arid grasses (Triodia R.Br.), highlighting our solutions to methodological challenges in the use of GBS data. We merged overlapping paired-end reads then optimised locus assembly in the program PyRAD to generate GBS data sets for phylogenetic and distance-based analyses. In addition to traditional concatenation analyses in RAxML, we also demonstrate the novel use of summary species tree analyses (taking gene trees as input) with GBS loci. We found that while species tree analyses were relatively robust to variation in PyRAD assembly parameters, our RAxML analyses resulted in well-supported but conflicting topologies under different assembly settings. Despite this conflict, multiple clades in the complex were consistently supported as distinct across analyses. Our GBS data assembly and analyses improve the resolution of taxa and phylogenetic relationships in the Triodia basedowii complex compared to our previous study based on Sanger sequencing of nuclear (ITS/ETS) and chloroplast (rps16-trnK spacer) markers. The genomic results also partly support previous evidence for hybridization between species in the complex. Our methodological insights for analysing GBS data will assist researchers using similar data to resolve phylogenetic relationships within species complexes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
