

A Critical Role for Astrocytes in Hypercapnic Vasodilation in Brain
- PMID: 28137973
- PMCID: PMC5354350
- DOI: 10.1523/JNEUROSCI.0005-16.2016
A Critical Role for Astrocytes in Hypercapnic Vasodilation in Brain
Erratum in
-
Erratum: Howarth et al., "A Critical Role for Astrocytes in Hypercapnic Vasodilation in Brain".J Neurosci. 2017 May 3;37(18):4860. doi: 10.1523/JNEUROSCI.1020-17.2017. J Neurosci. 2017. PMID: 28469012 Free PMC article. No abstract available.
Abstract
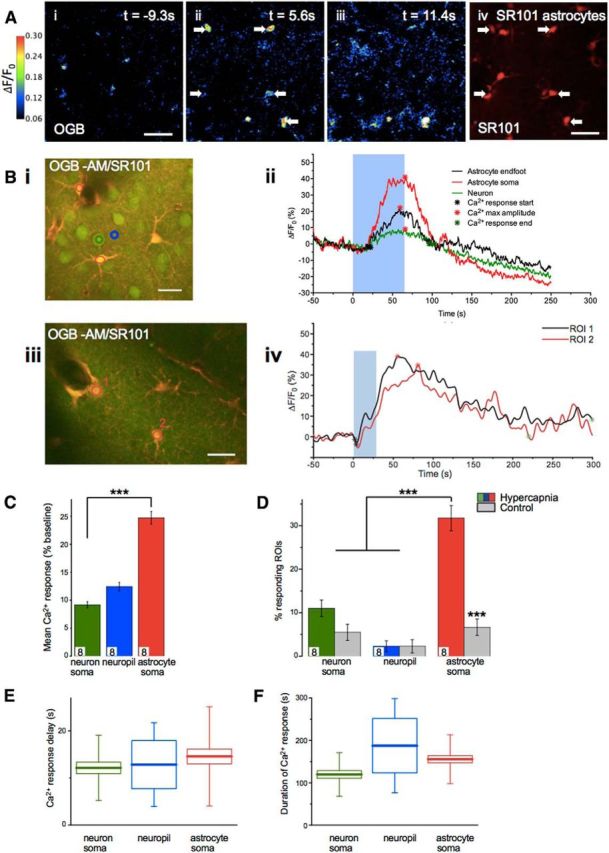
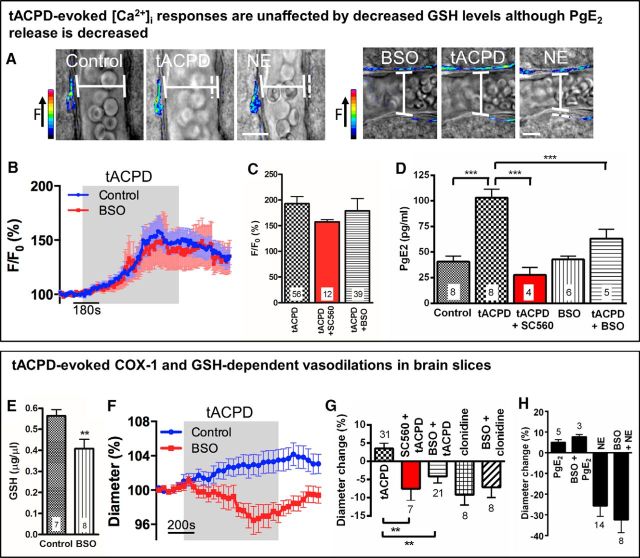
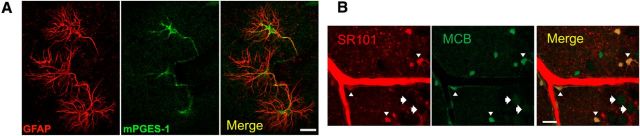
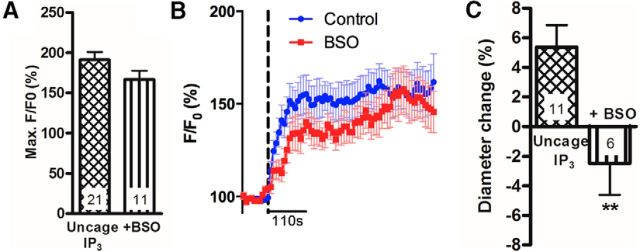
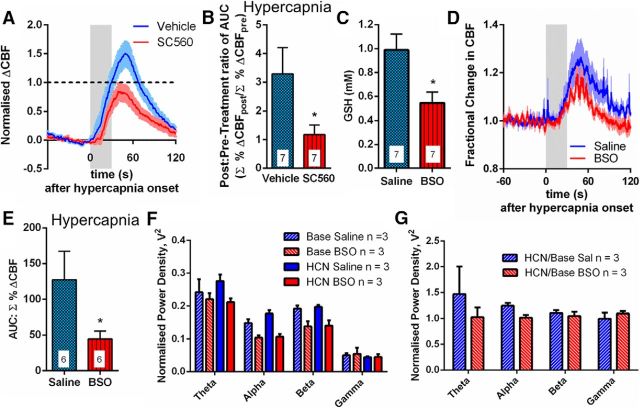
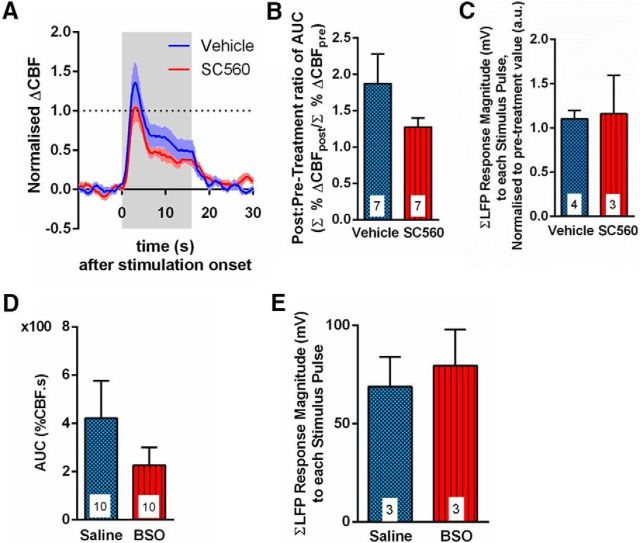
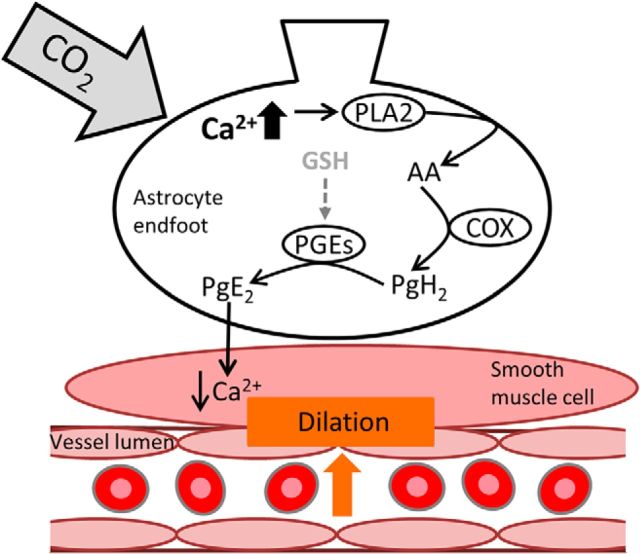
Cerebral blood flow (CBF) is controlled by arterial blood pressure, arterial CO2, arterial O2, and brain activity and is largely constant in the awake state. Although small changes in arterial CO2 are particularly potent to change CBF (1 mmHg variation in arterial CO2 changes CBF by 3%-4%), the coupling mechanism is incompletely understood. We tested the hypothesis that astrocytic prostaglandin E2 (PgE2) plays a key role for cerebrovascular CO2 reactivity, and that preserved synthesis of glutathione is essential for the full development of this response. We combined two-photon imaging microscopy in brain slices with in vivo work in rats and C57BL/6J mice to examine the hemodynamic responses to CO2 and somatosensory stimulation before and after inhibition of astrocytic glutathione and PgE2 synthesis. We demonstrate that hypercapnia (increased CO2) evokes an increase in astrocyte [Ca2+]i and stimulates COX-1 activity. The enzyme downstream of COX-1 that synthesizes PgE2 (microsomal prostaglandin E synthase-1) depends critically for its vasodilator activity on the level of glutathione in the brain. We show that, when glutathione levels are reduced, astrocyte calcium-evoked release of PgE2 is decreased and vasodilation triggered by increased astrocyte [Ca2+]iin vitro and by hypercapnia in vivo is inhibited. Astrocyte synthetic pathways, dependent on glutathione, are involved in cerebrovascular reactivity to CO2 Reductions in glutathione levels in aging, stroke, or schizophrenia could lead to dysfunctional regulation of CBF and subsequent neuronal damage.SIGNIFICANCE STATEMENT Neuronal activity leads to the generation of CO2, which has previously been shown to evoke cerebral blood flow (CBF) increases via the release of the vasodilator PgE2 We demonstrate that hypercapnia (increased CO2) evokes increases in astrocyte calcium signaling, which in turn stimulates COX-1 activity and generates downstream PgE2 production. We demonstrate that astrocyte calcium-evoked production of the vasodilator PgE2 is critically dependent on brain levels of the antioxidant glutathione. These data suggest a novel role for astrocytes in the regulation of CO2-evoked CBF responses. Furthermore, these results suggest that depleted glutathione levels, which occur in aging and stroke, will give rise to dysfunctional CBF regulation and may result in subsequent neuronal damage.
Keywords: astrocyte; calcium; cerebral blood flow; glutathione; hypercapnia.
Copyright © 2017 Howarth, Sutherland et al.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous