

Ganglionic GFAP + glial Gq-GPCR signaling enhances heart functions in vivo
- PMID: 28138563
- PMCID: PMC5256145
- DOI: 10.1172/jci.insight.90565
Ganglionic GFAP + glial Gq-GPCR signaling enhances heart functions in vivo
Abstract
The sympathetic nervous system (SNS) accelerates heart rate, increases cardiac contractility, and constricts resistance vessels. The activity of SNS efferent nerves is generated by a complex neural network containing neurons and glia. Gq G protein-coupled receptor (Gq-GPCR) signaling in glial fibrillary acidic protein-expressing (GFAP+) glia in the central nervous system supports neuronal function and regulates neuronal activity. It is unclear how Gq-GPCR signaling in GFAP+ glia affects the activity of sympathetic neurons or contributes to SNS-regulated cardiovascular functions. In this study, we investigated whether Gq-GPCR activation in GFAP+ glia modulates the regulatory effect of the SNS on the heart; transgenic mice expressing Gq-coupled DREADD (designer receptors exclusively activated by designer drugs) (hM3Dq) selectively in GFAP+ glia were used to address this question in vivo. We found that acute Gq-GPCR activation in peripheral GFAP+ glia significantly accelerated heart rate and increased left ventricle contraction. Pharmacological experiments suggest that the glial-induced cardiac changes were due to Gq-GPCR activation in satellite glial cells within the sympathetic ganglion; this activation led to increased norepinephrine (NE) release and beta-1 adrenergic receptor activation within the heart. Chronic glial Gq-GPCR activation led to hypotension in female Gfap-hM3Dq mice. This study provides direct evidence that Gq-GPCR activation in peripheral GFAP+ glia regulates cardiovascular functions in vivo.
Conflict of interest statement
The authors have declared that no conflict of interest exists.
Figures
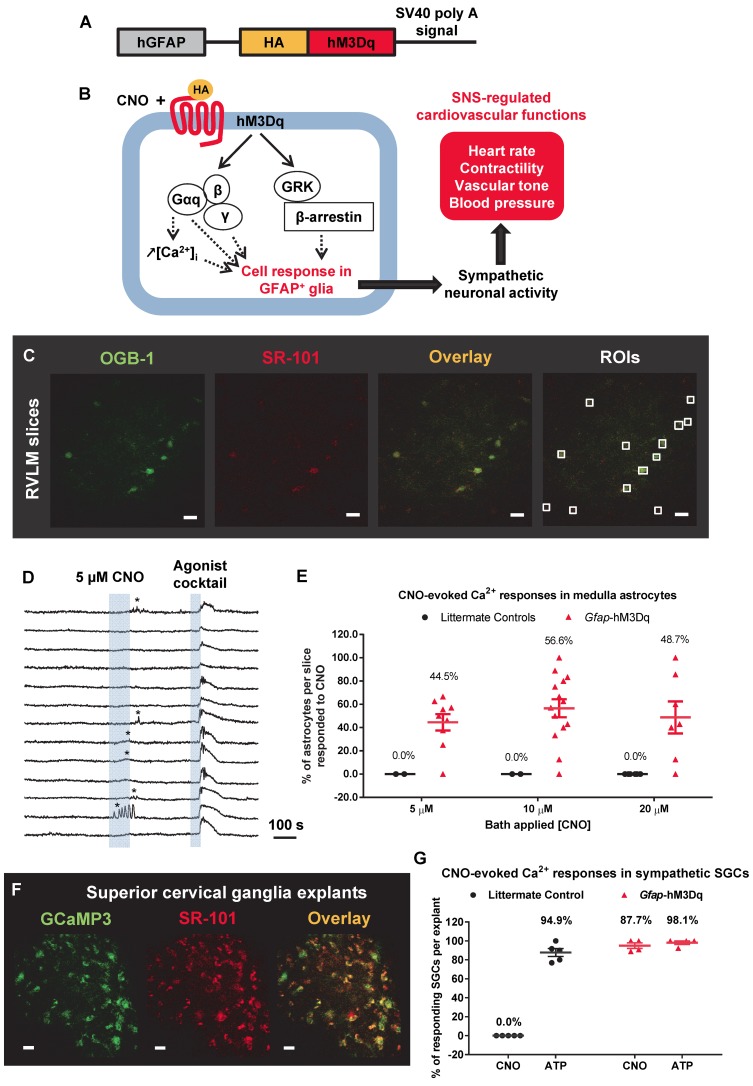
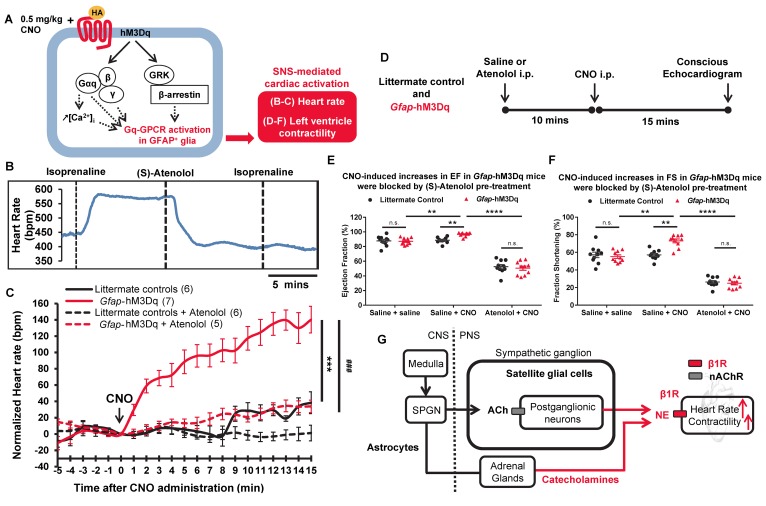
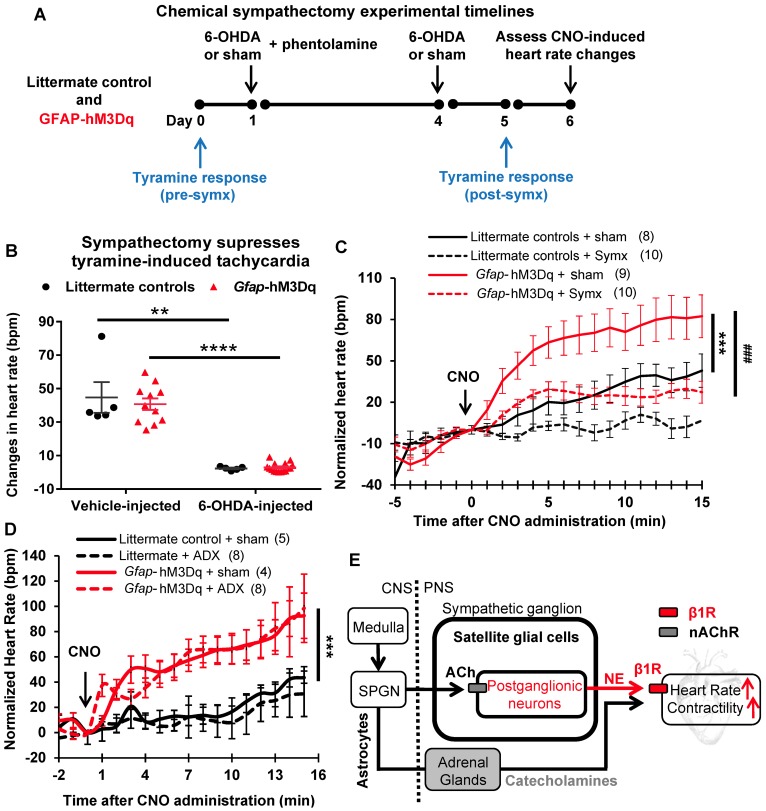
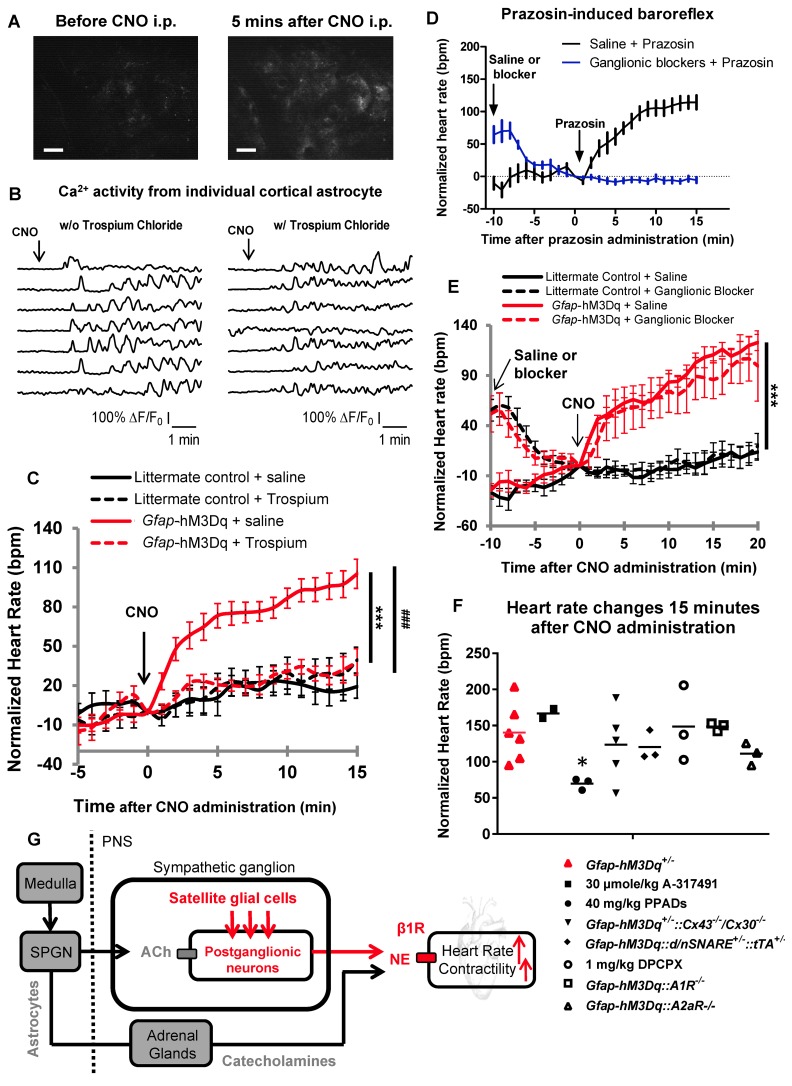
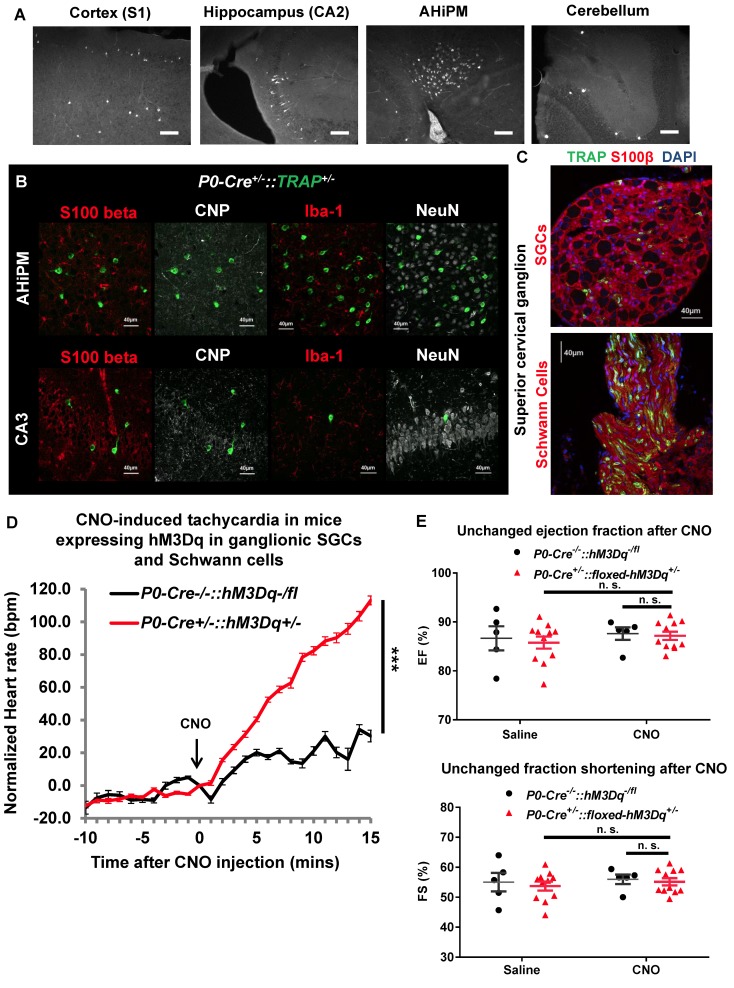
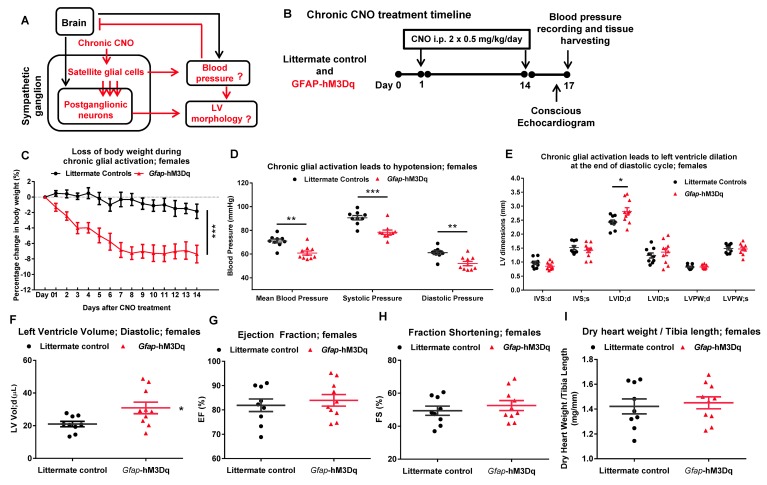
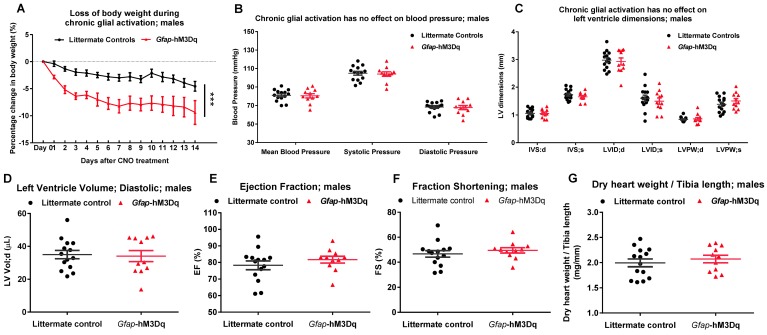
References
-
- Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7(5):335–346. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
