

Neonatal nonepileptic myoclonus is a prominent clinical feature of KCNQ2 gain-of-function variants R201C and R201H
- PMID: 28139826
- PMCID: PMC5339037
- DOI: 10.1111/epi.13676
Neonatal nonepileptic myoclonus is a prominent clinical feature of KCNQ2 gain-of-function variants R201C and R201H
Abstract
Objective: To analyze whether KCNQ2 R201C and R201H variants, which show atypical gain-of-function electrophysiologic properties in vitro, have a distinct clinical presentation and outcome.
Methods: Ten children with heterozygous, de novo KCNQ2 R201C or R201H variants were identified worldwide, using an institutional review board (IRB)-approved KCNQ2 patient registry and database. We reviewed medical records and, where possible, interviewed parents and treating physicians using a structured, detailed phenotype inventory focusing on the neonatal presentation and subsequent course.
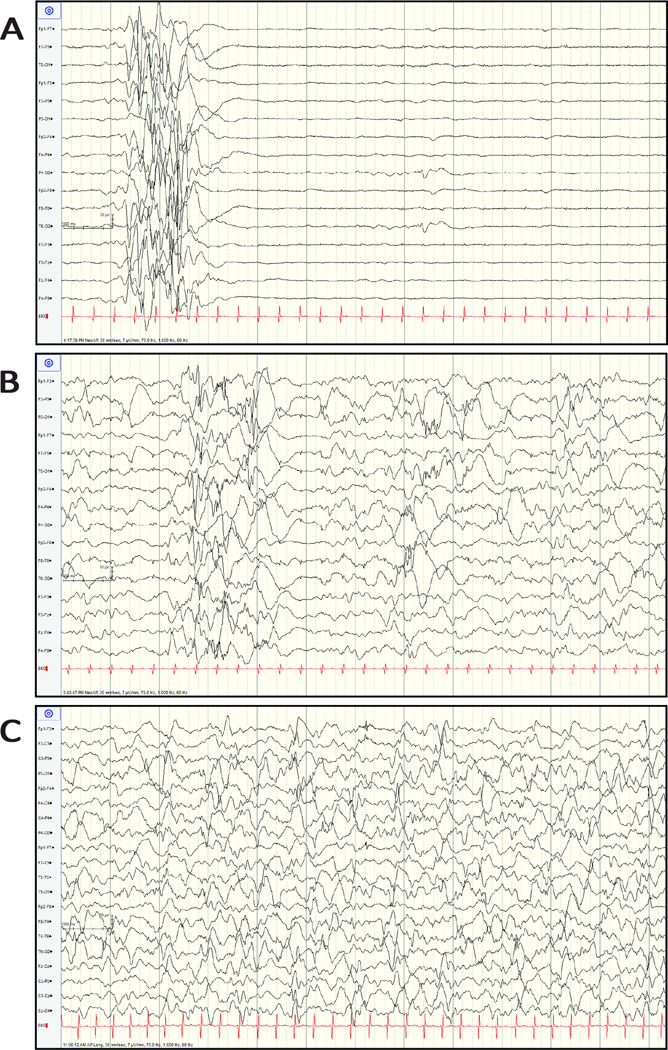
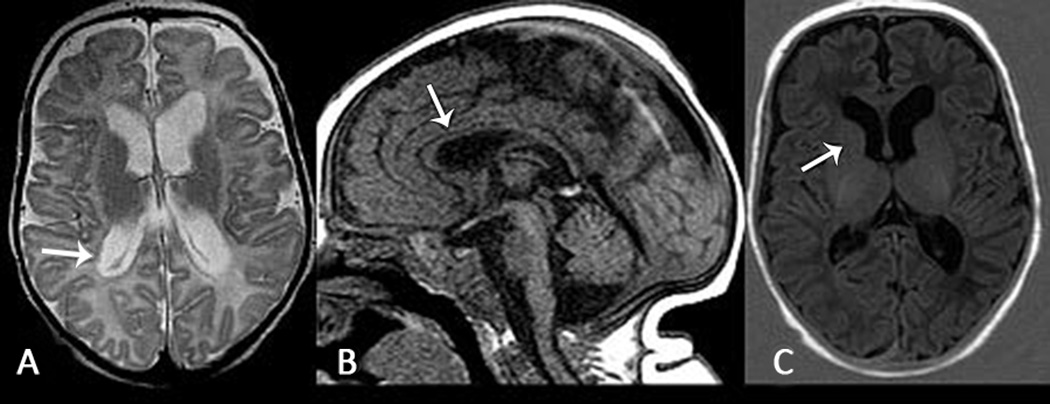
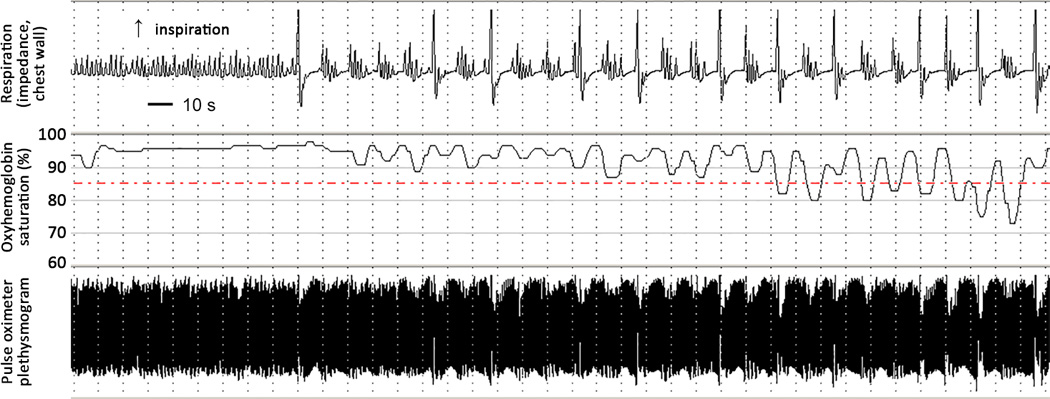
Results: Nine patients had encephalopathy from birth and presented with prominent startle-like myoclonus, which could be triggered by sound or touch. In seven patients, electroencephalography (EEG) was performed in the neonatal period and showed a burst-suppression pattern. However, myoclonus did not have an EEG correlate. In many patients the paroxysmal movements were misdiagnosed as seizures. Seven patients developed epileptic spasms in infancy. In all patients, EEG showed a slow background and multifocal epileptiform discharges later in life. Other prominent features included respiratory dysfunction (perinatal respiratory failure and/or chronic hypoventilation), hypomyelination, reduced brain volume, and profound developmental delay. One patient had a later onset, and sequencing indicated that a low abundance (~20%) R201C variant had arisen by postzygotic mosaicism.
Significance: Heterozygous KCNQ2 R201C and R201H gain-of-function variants present with profound neonatal encephalopathy in the absence of neonatal seizures. Neonates present with nonepileptic myoclonus that is often misdiagnosed and treated as seizures. Prognosis is poor. This clinical presentation is distinct from the phenotype associated with loss-of-function variants, supporting the value of in vitro functional screening. These findings suggest that gain-of-function and loss-of-function variants need different targeted therapeutic approaches.
Keywords: Epileptic encephalopathy; Infantile spasms; KCNQ2; Myoclonus; Neonatal seizures.
Wiley Periodicals, Inc. © 2017 International League Against Epilepsy.
Conflict of interest statement
The remaining authors have no conflicts of interest.
Figures
References
-
- Biervert C, Schroeder BC, Kubisch C, et al. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. - PubMed
-
- Grinton BE, Heron SE, Pelekanos JT, et al. Familial neonatal seizures in 36 families: Clinical and genetic features correlate with outcome. Epilepsia. 2015;56:1071–1080. - PubMed
-
- Singh NA, Charlier C, Stauffer D, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29. - PubMed
-
- Weckhuysen S, Mandelstam S, Suls A, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol. 2012;71:15–25. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
