

BRILIA: Integrated Tool for High-Throughput Annotation and Lineage Tree Assembly of B-Cell Repertoires
- PMID: 28144239
- PMCID: PMC5239784
- DOI: 10.3389/fimmu.2016.00681
BRILIA: Integrated Tool for High-Throughput Annotation and Lineage Tree Assembly of B-Cell Repertoires
Abstract
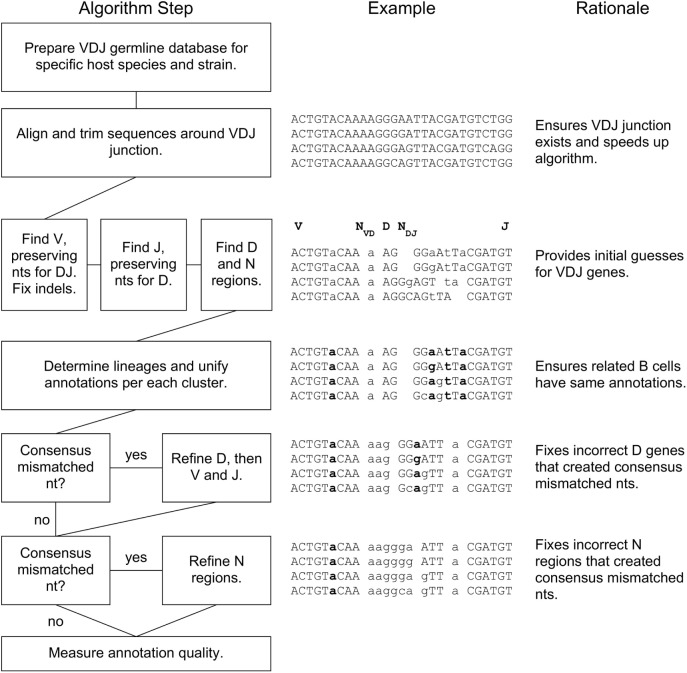
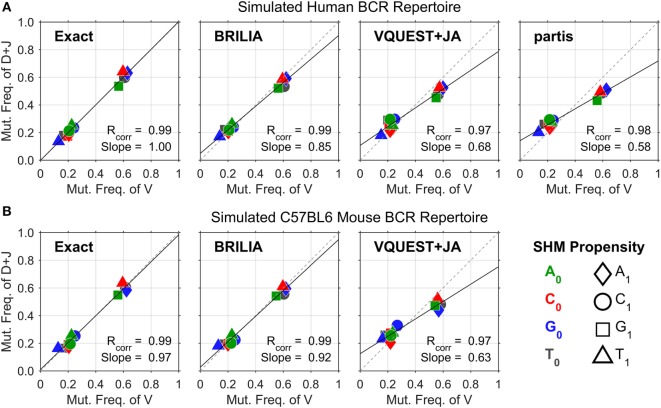
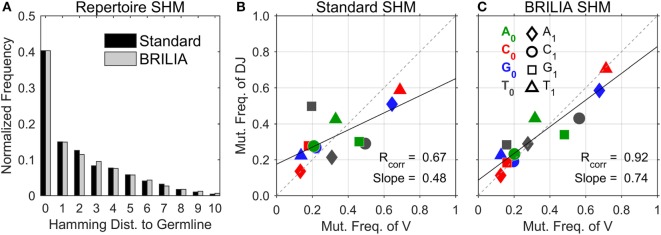
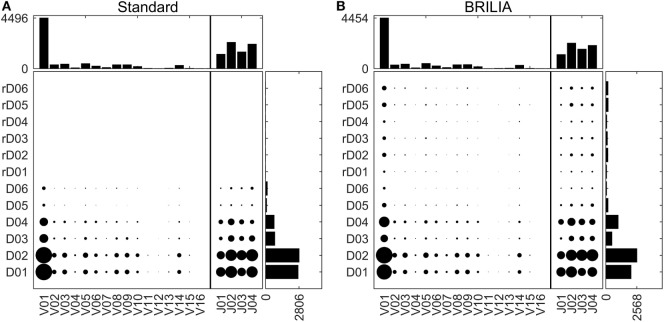
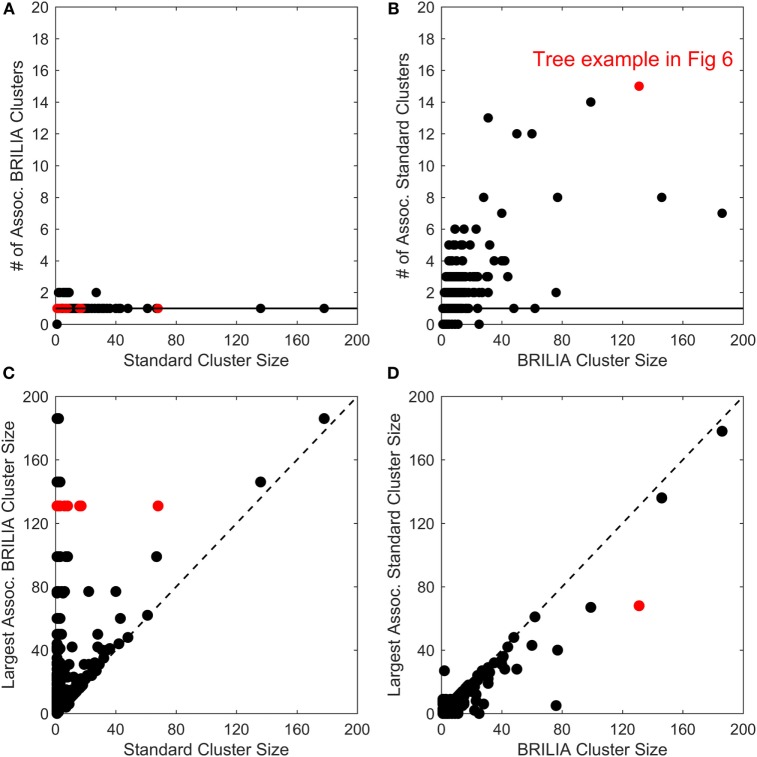
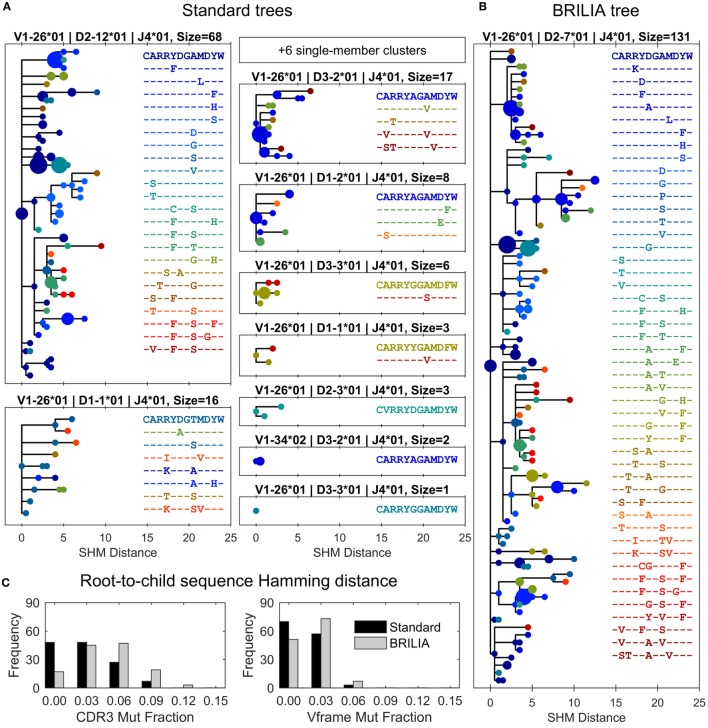
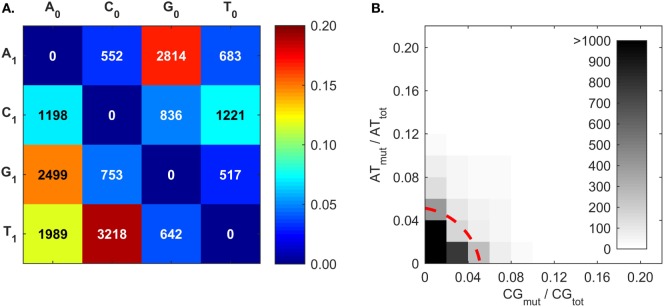
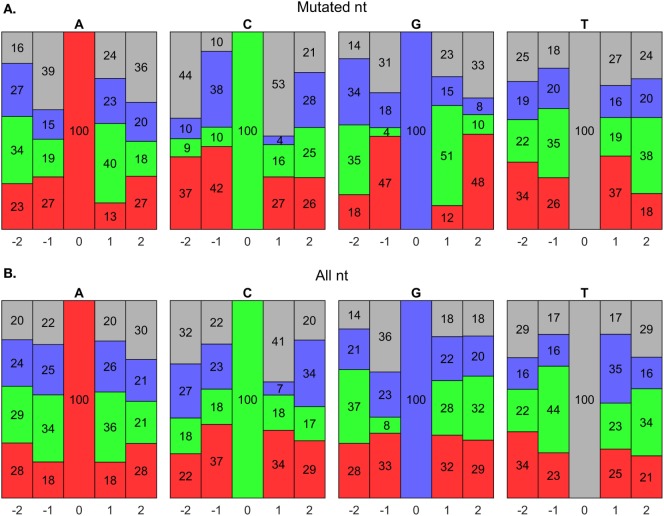
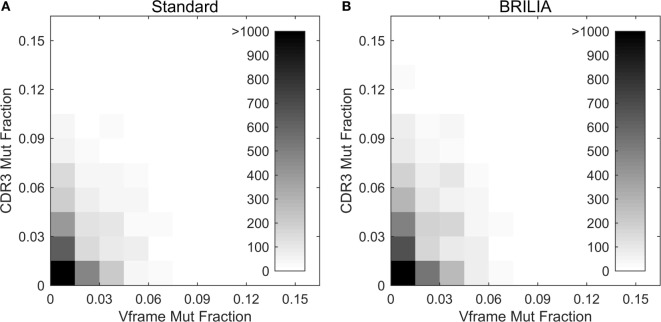
The somatic diversity of antigen-recognizing B-cell receptors (BCRs) arises from Variable (V), Diversity (D), and Joining (J) (VDJ) recombination and somatic hypermutation (SHM) during B-cell development and affinity maturation. The VDJ junction of the BCR heavy chain forms the highly variable complementarity determining region 3 (CDR3), which plays a critical role in antigen specificity and binding affinity. Tracking the selection and mutation of the CDR3 can be useful in characterizing humoral responses to infection and vaccination. Although tens to hundreds of thousands of unique BCR genes within an expressed B-cell repertoire can now be resolved with high-throughput sequencing, tracking SHMs is still challenging because existing annotation methods are often limited by poor annotation coverage, inconsistent SHM identification across the VDJ junction, or lack of B-cell lineage data. Here, we present B-cell repertoire inductive lineage and immunosequence annotator (BRILIA), an algorithm that leverages repertoire-wide sequencing data to globally improve the VDJ annotation coverage, lineage tree assembly, and SHM identification. On benchmark tests against simulated human and mouse BCR repertoires, BRILIA correctly annotated germline and clonally expanded sequences with 94 and 70% accuracy, respectively, and it has a 90% SHM-positive prediction rate in the CDR3 of heavily mutated sequences; these are substantial improvements over existing methods. We used BRILIA to process BCR sequences obtained from splenic germinal center B cells extracted from C57BL/6 mice. BRILIA returned robust B-cell lineage trees and yielded SHM patterns that are consistent across the VDJ junction and agree with known biological mechanisms of SHM. By contrast, existing BCR annotation tools, which do not account for repertoire-wide clonal relationships, systematically underestimated both the size of clonally related B-cell clusters and yielded inconsistent SHM frequencies. We demonstrate BRILIA's utility in B-cell repertoire studies related to VDJ gene usage, mechanisms for adenosine mutations, and SHM hot spot motifs. Furthermore, we show that the complete gene usage annotation and SHM identification across the entire CDR3 are essential for studying the B-cell affinity maturation process through immunosequencing methods.
Keywords: B-cell receptor (BCR); VDJ; annotation; lineage; repertoire; somatic hypermutation (SHM).
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources