

Computational methods in drug discovery
- PMID: 28144341
- PMCID: PMC5238551
- DOI: 10.3762/bjoc.12.267
Computational methods in drug discovery
Abstract
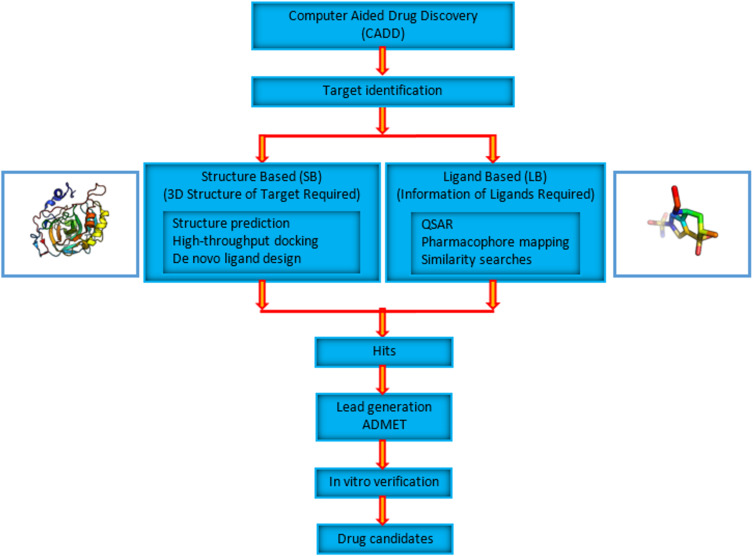
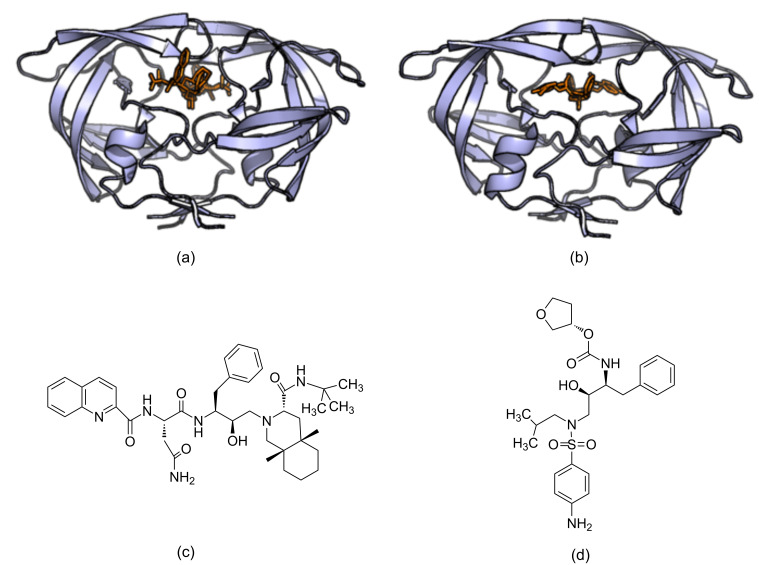
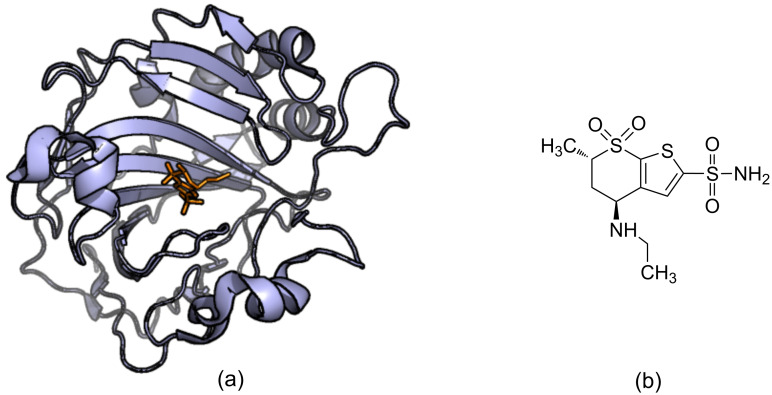
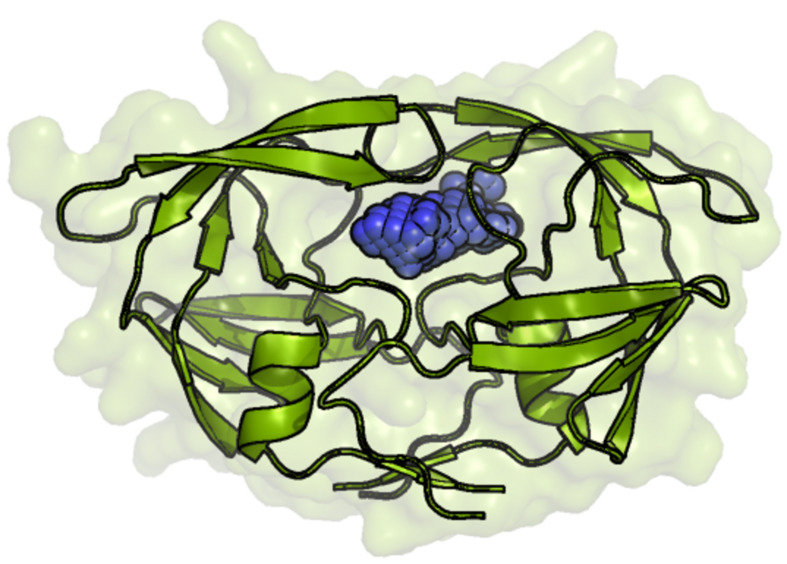
The process for drug discovery and development is challenging, time consuming and expensive. Computer-aided drug discovery (CADD) tools can act as a virtual shortcut, assisting in the expedition of this long process and potentially reducing the cost of research and development. Today CADD has become an effective and indispensable tool in therapeutic development. The human genome project has made available a substantial amount of sequence data that can be used in various drug discovery projects. Additionally, increasing knowledge of biological structures, as well as increasing computer power have made it possible to use computational methods effectively in various phases of the drug discovery and development pipeline. The importance of in silico tools is greater than ever before and has advanced pharmaceutical research. Here we present an overview of computational methods used in different facets of drug discovery and highlight some of the recent successes. In this review, both structure-based and ligand-based drug discovery methods are discussed. Advances in virtual high-throughput screening, protein structure prediction methods, protein-ligand docking, pharmacophore modeling and QSAR techniques are reviewed.
Keywords: ADME; LBDD; QSAR; SBDD; computer-aided drug design; docking; free energy; high-throughput screening; lead optimization; machine learning; pharmacophore; scoring; target flexibility.
Figures
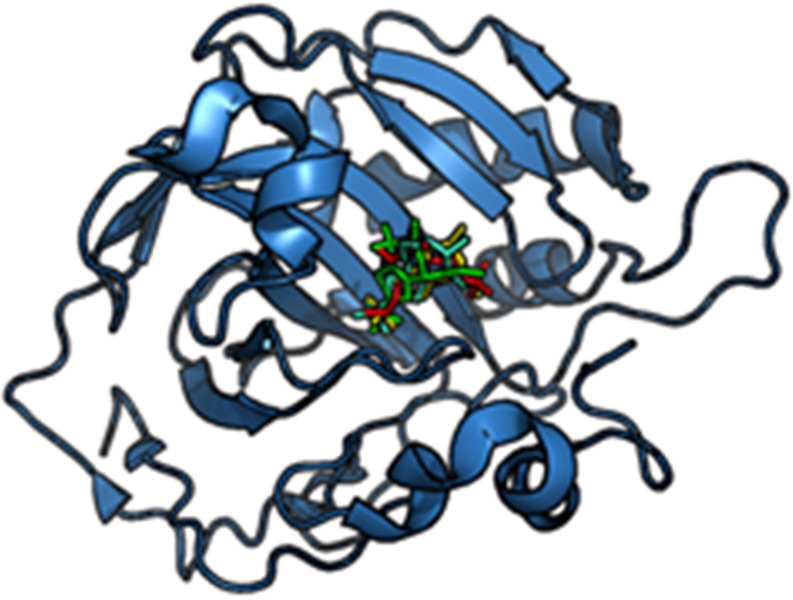
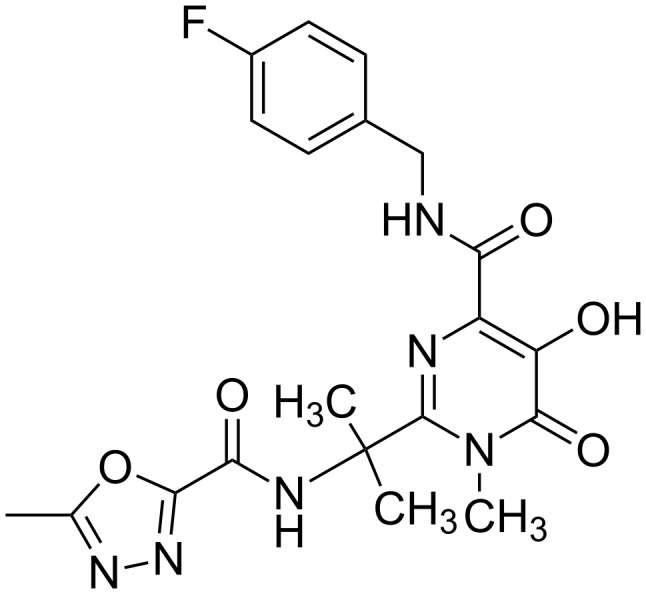
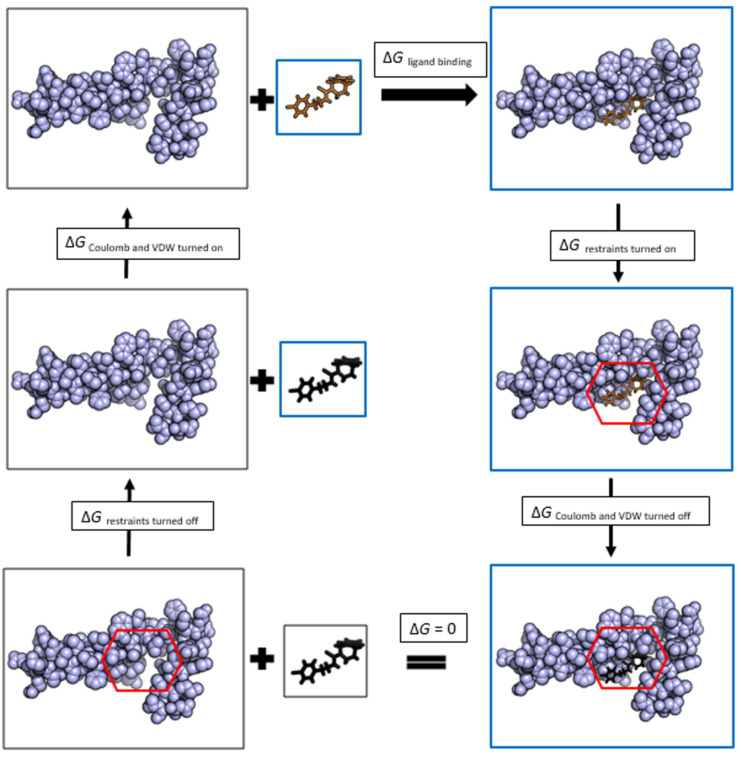
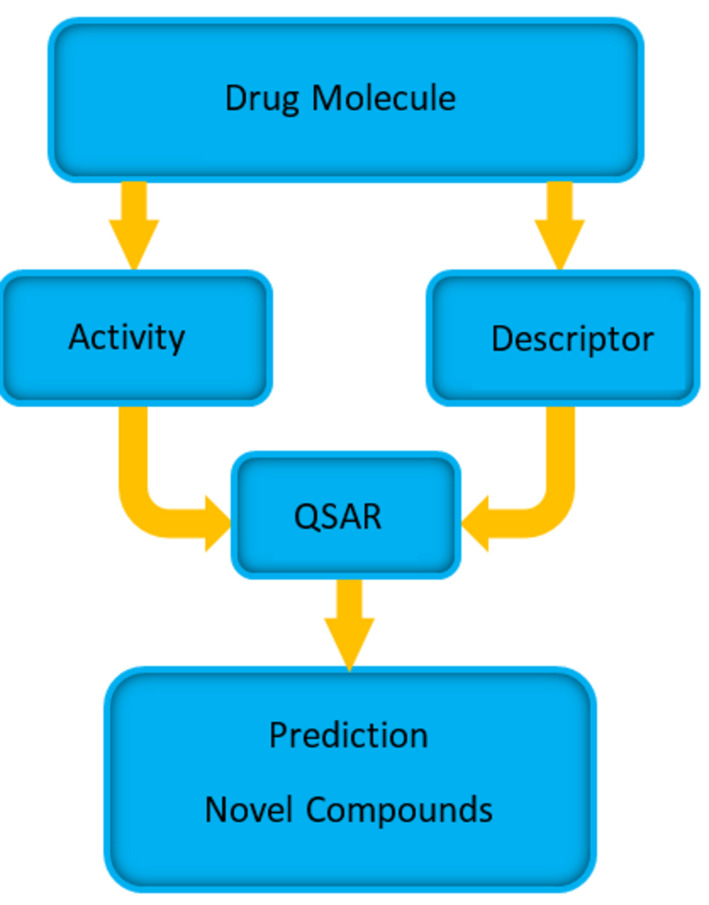
References
-
- Tollman P. A Revolution in R&D: How genomics and genetics are transforming the biopharmaceutical industry. 2001.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources