

Computational Prediction of the Heterodimeric and Higher-Order Structure of gpE1/gpE2 Envelope Glycoproteins Encoded by Hepatitis C Virus
- PMID: 28148799
- PMCID: PMC5375693
- DOI: 10.1128/JVI.02309-16
Computational Prediction of the Heterodimeric and Higher-Order Structure of gpE1/gpE2 Envelope Glycoproteins Encoded by Hepatitis C Virus
Abstract
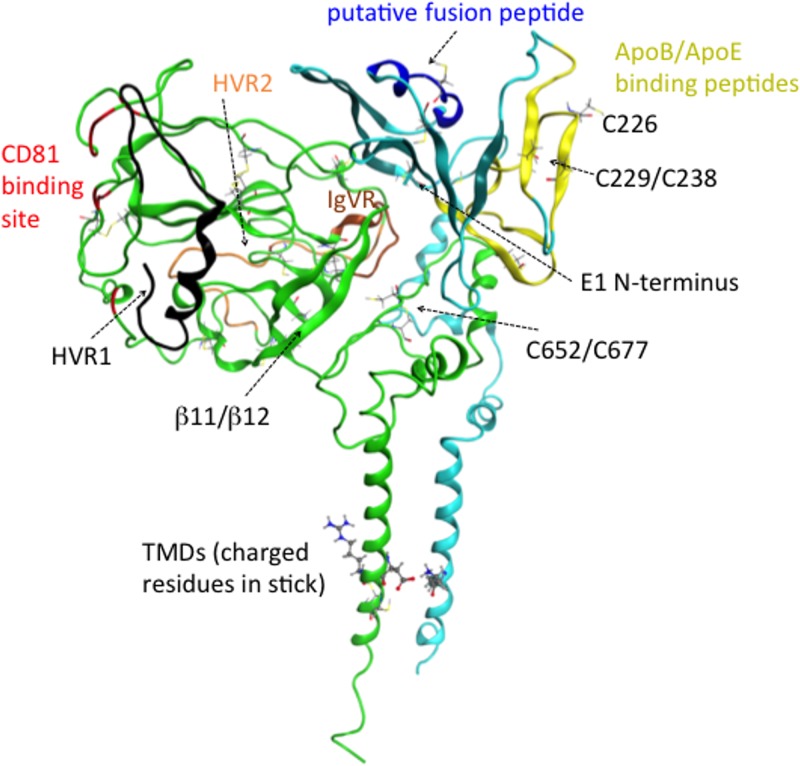
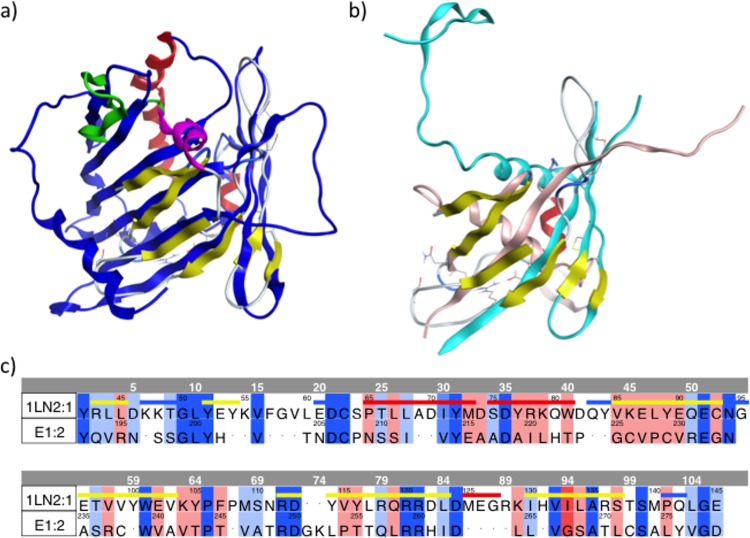
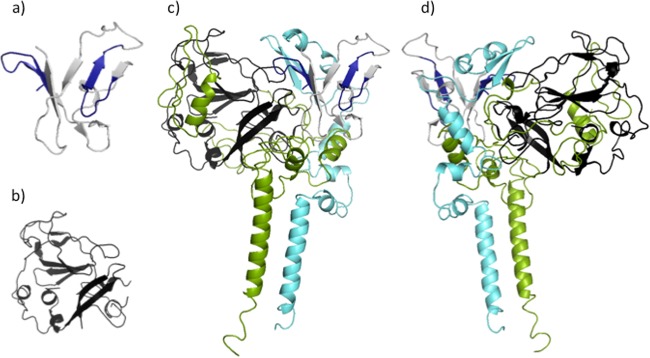
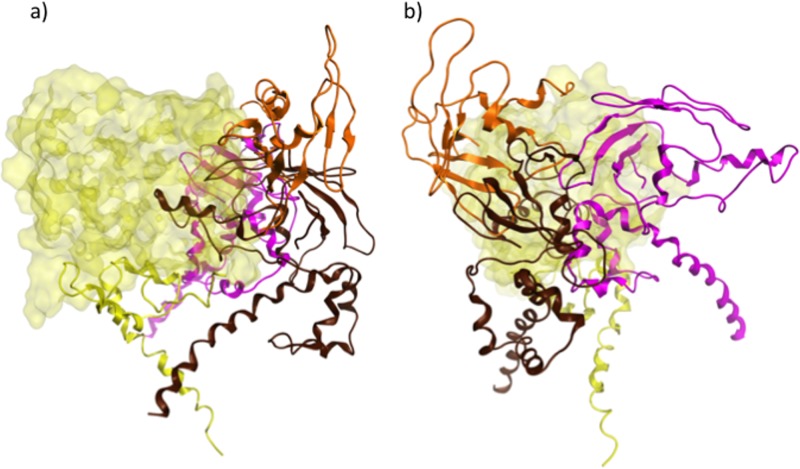
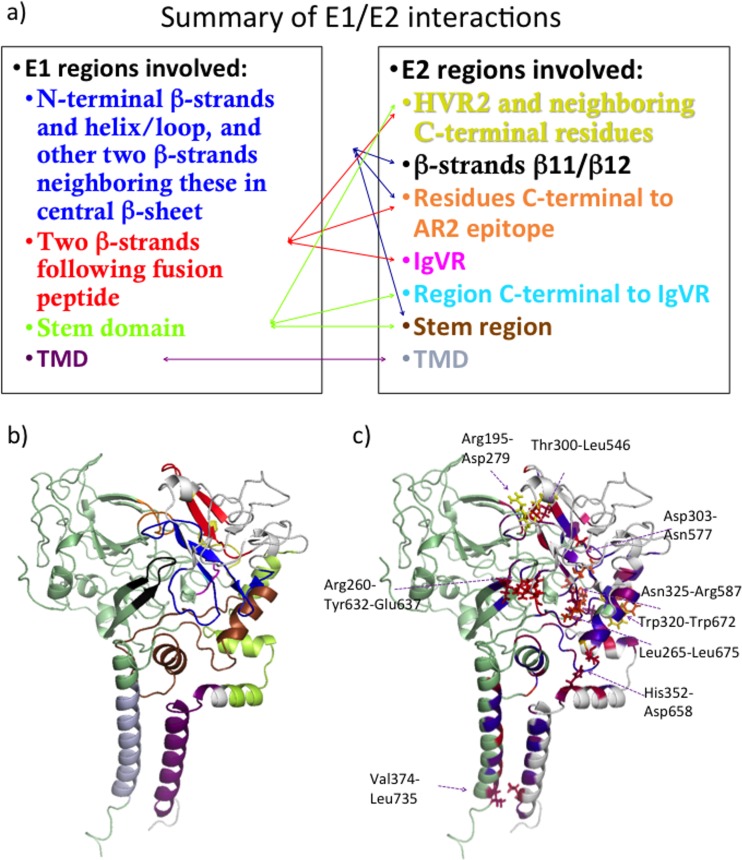
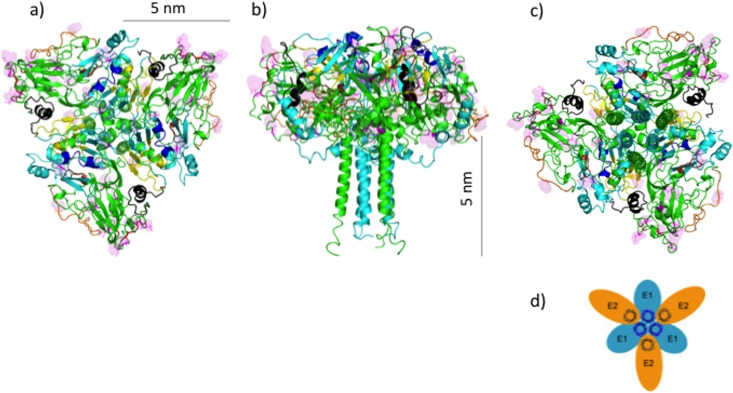
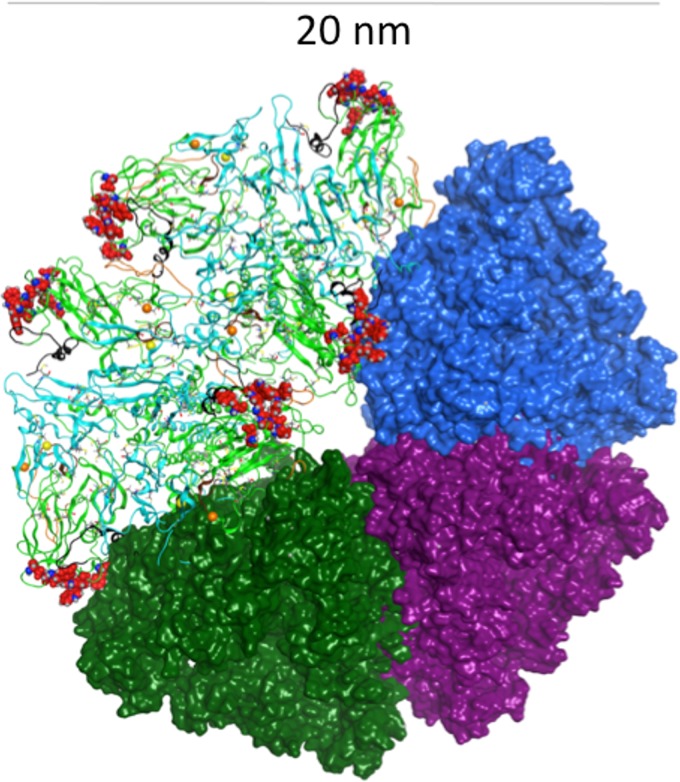
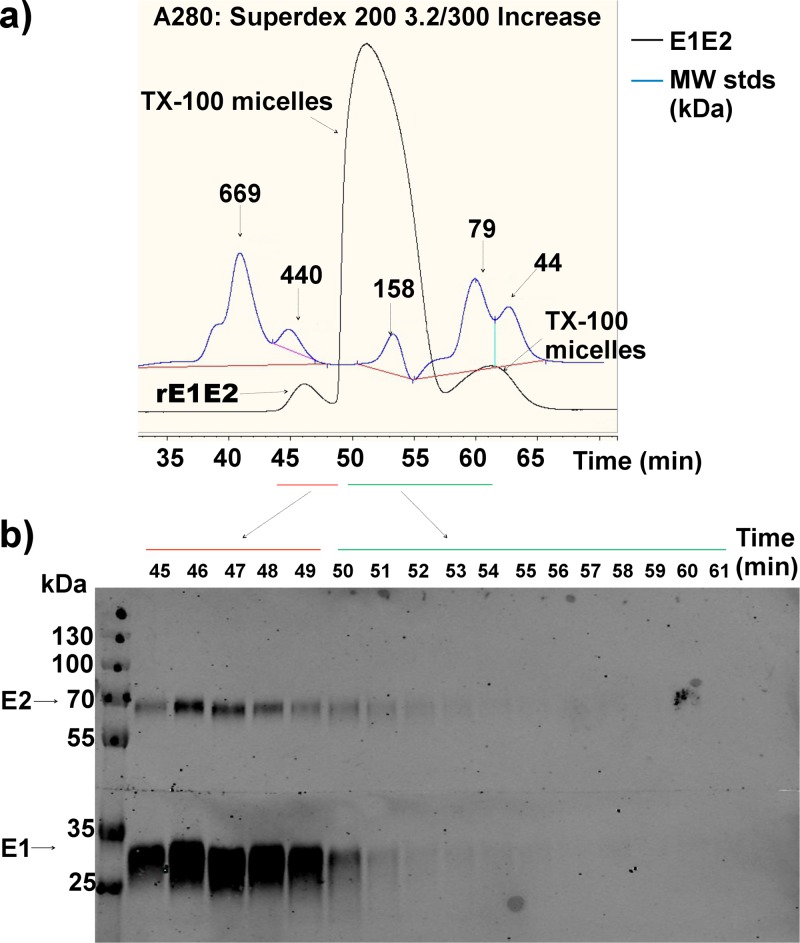
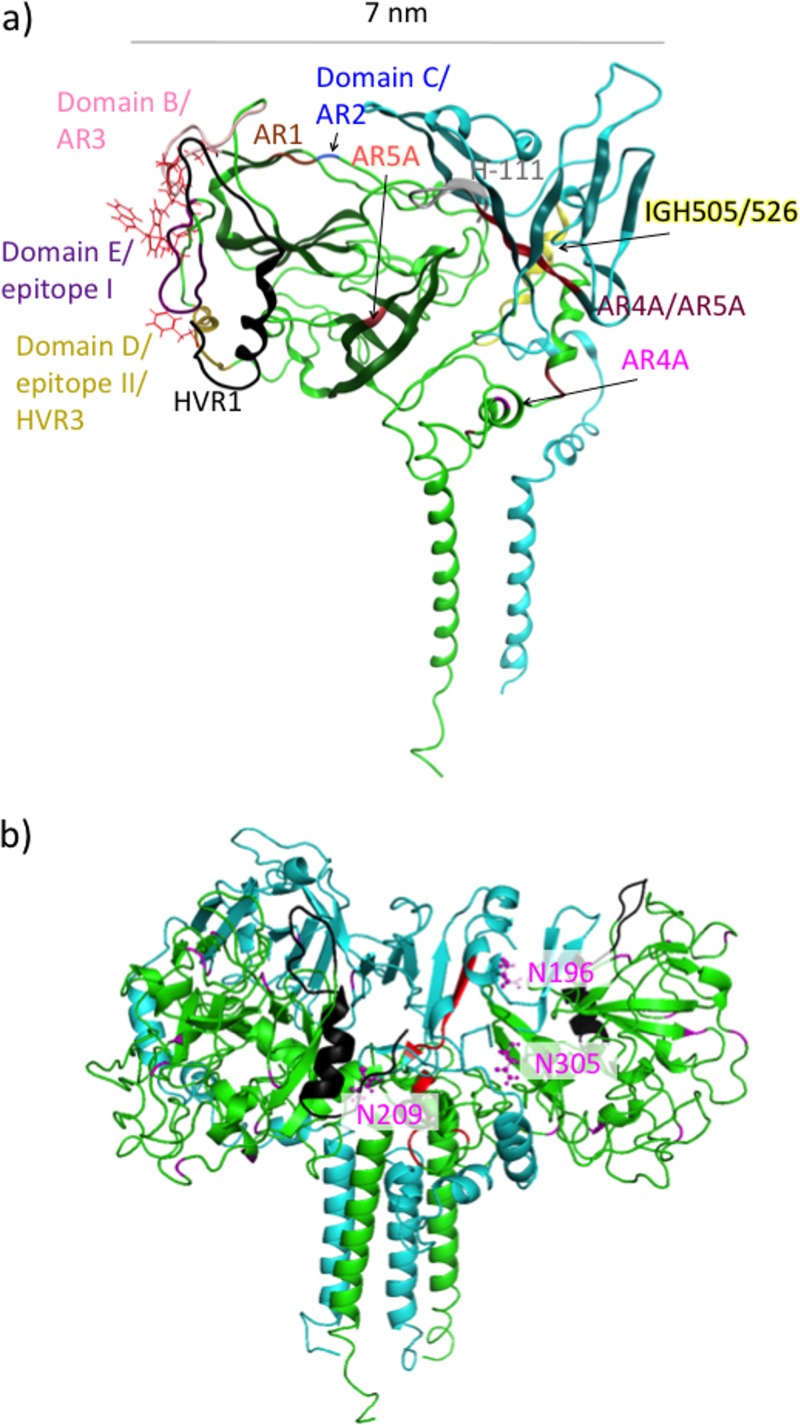
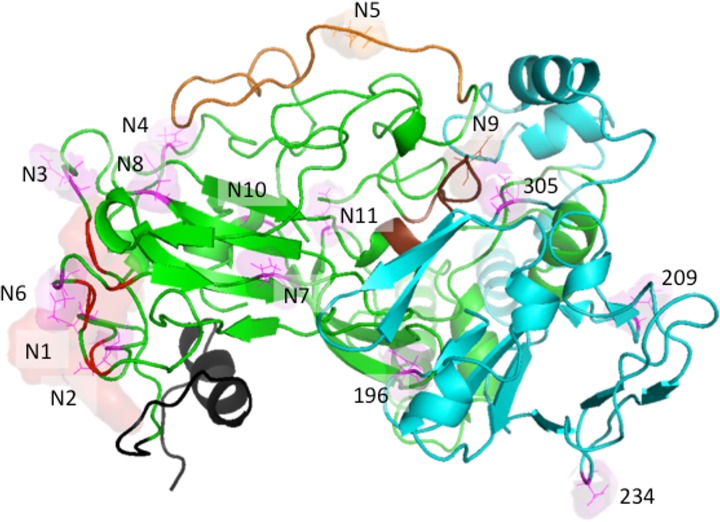
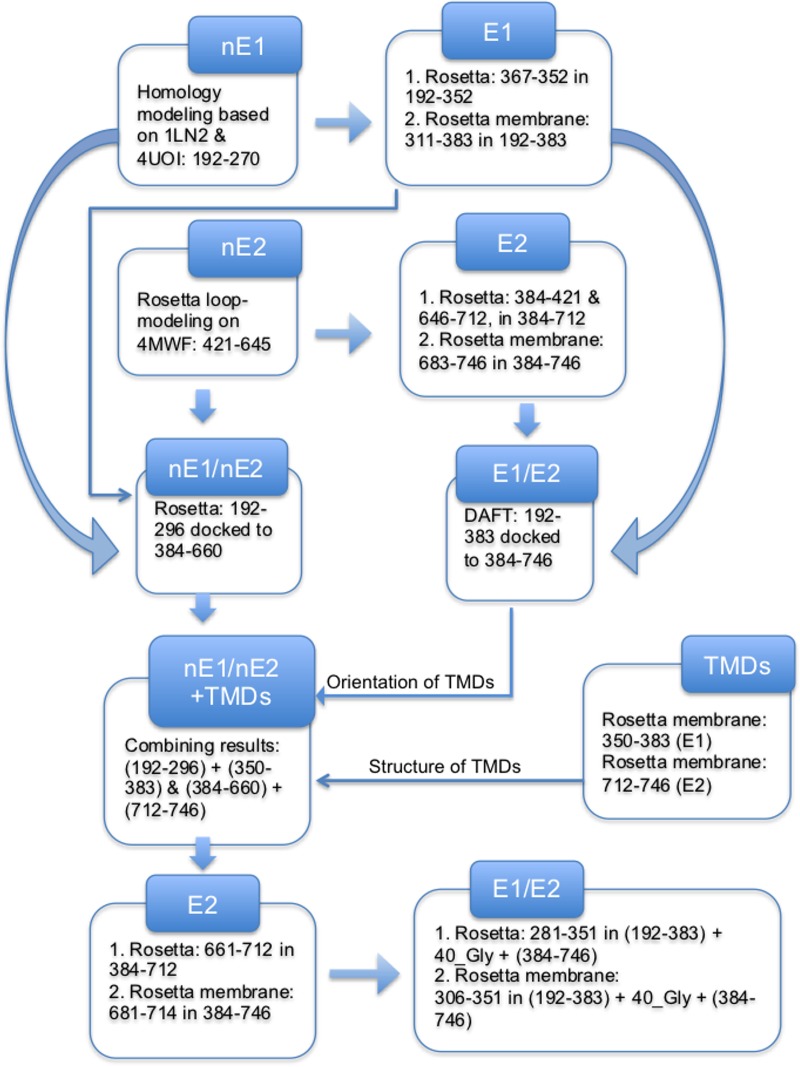
Despite the recent success of newly developed direct-acting antivirals against hepatitis C, the disease continues to be a global health threat due to the lack of diagnosis of most carriers and the high cost of treatment. The heterodimer formed by glycoproteins E1 and E2 within the hepatitis C virus (HCV) lipid envelope is a potential vaccine candidate and antiviral target. While the structure of E1/E2 has not yet been resolved, partial crystal structures of the E1 and E2 ectodomains have been determined. The unresolved parts of the structure are within the realm of what can be modeled with current computational modeling tools. Furthermore, a variety of additional experimental data is available to support computational predictions of E1/E2 structure, such as data from antibody binding studies, cryo-electron microscopy (cryo-EM), mutational analyses, peptide binding analysis, linker-scanning mutagenesis, and nuclear magnetic resonance (NMR) studies. In accordance with these rich experimental data, we have built an in silico model of the full-length E1/E2 heterodimer. Our model supports that E1/E2 assembles into a trimer, which was previously suggested from a study by Falson and coworkers (P. Falson, B. Bartosch, K. Alsaleh, B. A. Tews, A. Loquet, Y. Ciczora, L. Riva, C. Montigny, C. Montpellier, G. Duverlie, E. I. Pecheur, M. le Maire, F. L. Cosset, J. Dubuisson, and F. Penin, J. Virol. 89:10333-10346, 2015, https://doi.org/10.1128/JVI.00991-15). Size exclusion chromatography and Western blotting data obtained by using purified recombinant E1/E2 support our hypothesis. Our model suggests that during virus assembly, the trimer of E1/E2 may be further assembled into a pentamer, with 12 pentamers comprising a single HCV virion. We anticipate that this new model will provide a useful framework for HCV envelope structure and the development of antiviral strategies.IMPORTANCE One hundred fifty million people have been estimated to be infected with hepatitis C virus, and many more are at risk for infection. A better understanding of the structure of the HCV envelope, which is responsible for attachment and fusion, could aid in the development of a vaccine and/or new treatments for this disease. We draw upon computational techniques to predict a full-length model of the E1/E2 heterodimer based on the partial crystal structures of the envelope glycoproteins E1 and E2. E1/E2 has been widely studied experimentally, and this provides valuable data, which has assisted us in our modeling. Our proposed structure is used to suggest the organization of the HCV envelope. We also present new experimental data from size exclusion chromatography that support our computational prediction of a trimeric oligomeric state of E1/E2.
Keywords: E1/E2 heterodimer; Martini force field; Rosetta program; computational structural biology; hepatitis C virus.
Copyright © 2017 American Society for Microbiology.
Figures
References
-
- Thomssen R, Bonk S, Thiele A. 1993. Density heterogeneities of hepatitis C virus in human sera due to the binding of beta-lipoproteins and immunoglobulins. Med Microbiol Immunol 182:329–334. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
