

The E3 ligase Mule protects the heart against oxidative stress and mitochondrial dysfunction through Myc-dependent inactivation of Pgc-1α and Pink1
- PMID: 28148912
- PMCID: PMC5288653
- DOI: 10.1038/srep41490
The E3 ligase Mule protects the heart against oxidative stress and mitochondrial dysfunction through Myc-dependent inactivation of Pgc-1α and Pink1
Abstract
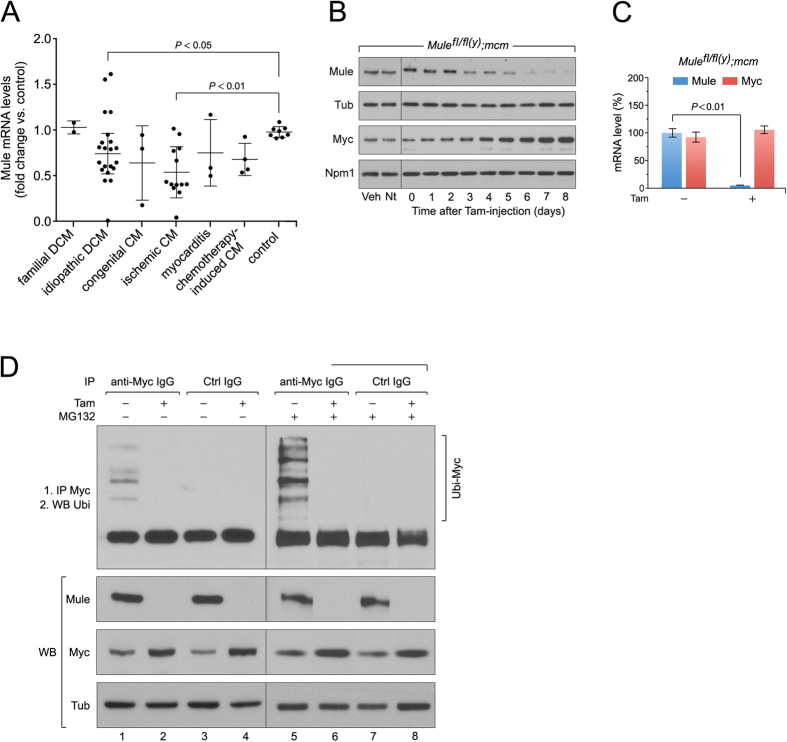
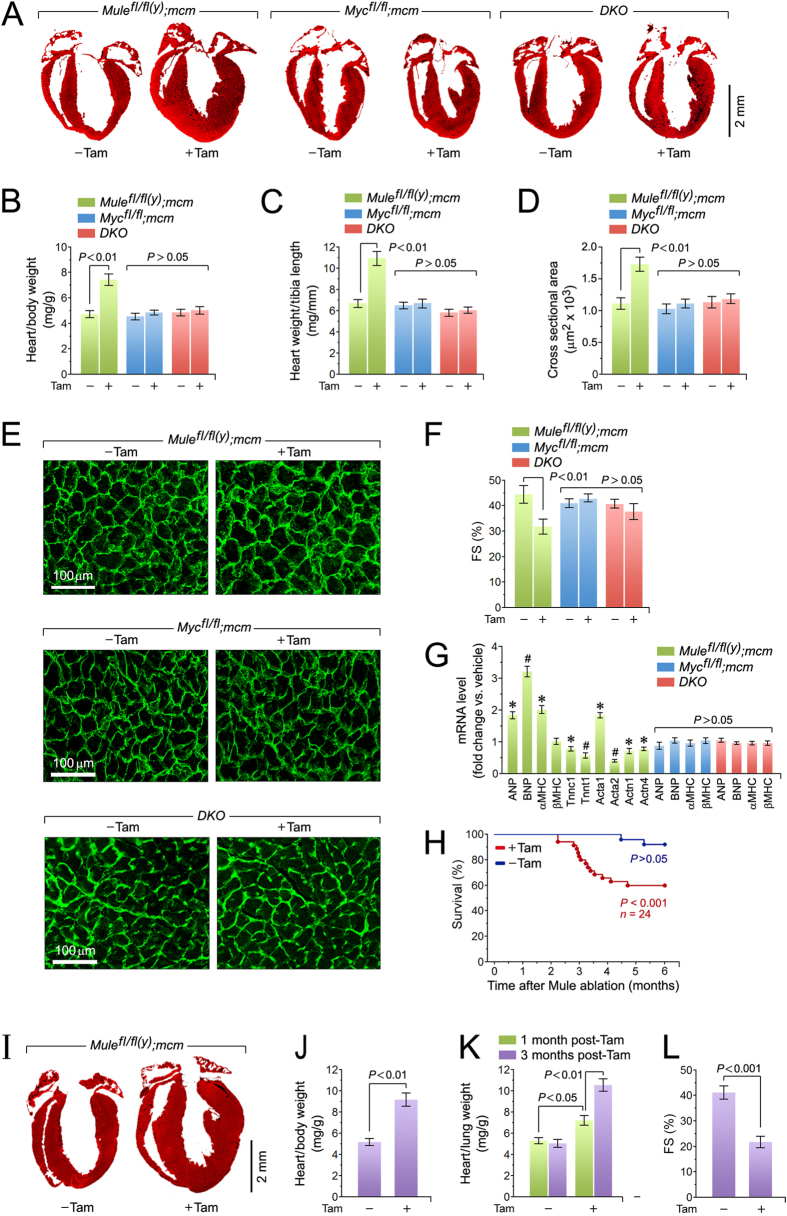
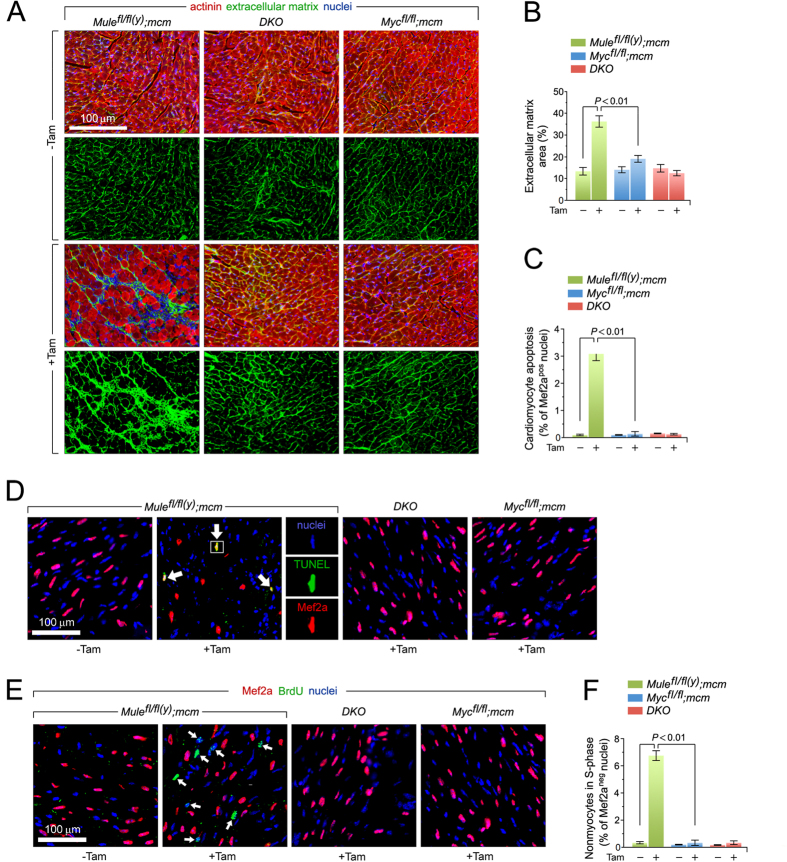
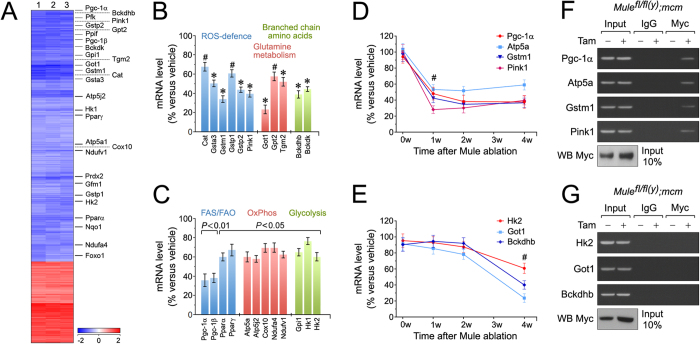
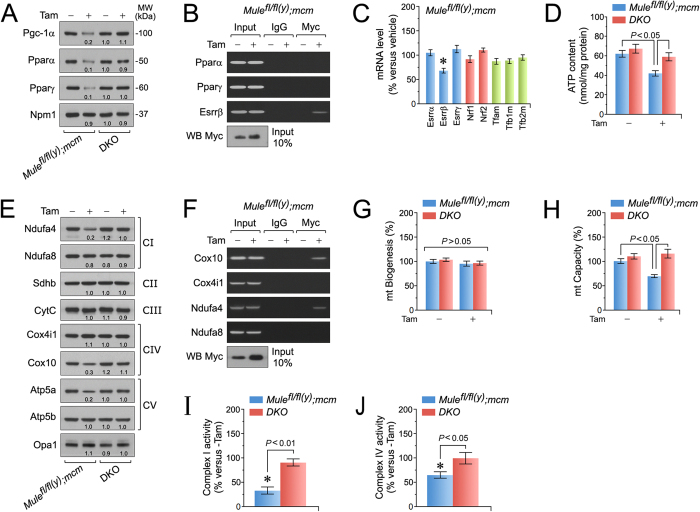
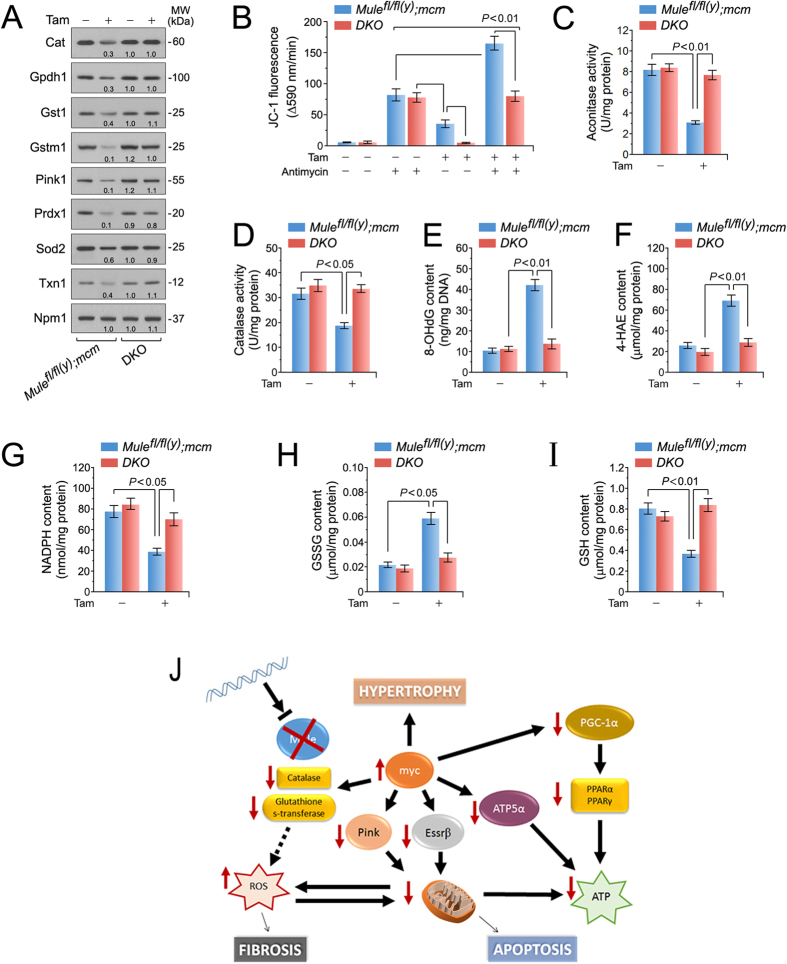
Cardiac homeostasis requires proper control of protein turnover. Protein degradation is principally controlled by the Ubiquitin-Proteasome System. Mule is an E3 ubiquitin ligase that regulates cellular growth, DNA repair and apoptosis to maintain normal tissue architecture. However, Mule's function in the heart has yet to be described. In a screen, we found reduced Mule expression in left ventricular samples from end-stage heart failure patients. Consequently, we generated conditional cardiac-specific Mule knockout (Mule fl/fl(y);mcm) mice. Mule ablation in adult Mule fl/fl(y);mcm mice prevented myocardial c-Myc polyubiquitination, leading to c-Myc accumulation and subsequent reduced expression of Pgc-1α, Pink1, and mitochondrial complex proteins. Furthermore, these mice developed spontaneous cardiac hypertrophy, left ventricular dysfunction, and early mortality. Co-deletion of Mule and c-Myc rescued this phenotype. Our data supports an indispensable role for Mule in cardiac homeostasis through the regulation of mitochondrial function via maintenance of Pgc-1α and Pink1 expression and persistent negative regulation of c-Myc.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Similar articles
-
Cardiac-specific ablation of the E3 ubiquitin ligase Mdm2 leads to oxidative stress, broad mitochondrial deficiency and early death.PLoS One. 2017 Dec 21;12(12):e0189861. doi: 10.1371/journal.pone.0189861. eCollection 2017. PLoS One. 2017. PMID: 29267372 Free PMC article.
-
Inactivation of EWS reduces PGC-1α protein stability and mitochondrial homeostasis.Proc Natl Acad Sci U S A. 2015 May 12;112(19):6074-9. doi: 10.1073/pnas.1504391112. Epub 2015 Apr 27. Proc Natl Acad Sci U S A. 2015. PMID: 25918410 Free PMC article.
-
Deubiquitinating enzyme CYLD mediates pressure overload-induced cardiac maladaptive remodeling and dysfunction via downregulating Nrf2.J Mol Cell Cardiol. 2015 Jul;84:143-53. doi: 10.1016/j.yjmcc.2015.04.012. Epub 2015 Apr 30. J Mol Cell Cardiol. 2015. PMID: 25935309
-
Perturbations in the gene regulatory pathways controlling mitochondrial energy production in the failing heart.Biochim Biophys Acta. 2013 Apr;1833(4):840-7. doi: 10.1016/j.bbamcr.2012.08.015. Epub 2012 Aug 31. Biochim Biophys Acta. 2013. PMID: 22964268 Free PMC article. Review.
-
Nix Nought Nothing: fairy tale or real deal.J Mol Cell Cardiol. 2011 Oct;51(4):497-500. doi: 10.1016/j.yjmcc.2010.09.011. Epub 2010 Sep 18. J Mol Cell Cardiol. 2011. PMID: 20858501 Free PMC article. Review.
Cited by
-
Cardiac-specific ablation of the E3 ubiquitin ligase Mdm2 leads to oxidative stress, broad mitochondrial deficiency and early death.PLoS One. 2017 Dec 21;12(12):e0189861. doi: 10.1371/journal.pone.0189861. eCollection 2017. PLoS One. 2017. PMID: 29267372 Free PMC article.
-
Targeting MCL-1 protein to treat cancer: opportunities and challenges.Front Oncol. 2023 Jul 31;13:1226289. doi: 10.3389/fonc.2023.1226289. eCollection 2023. Front Oncol. 2023. PMID: 37601693 Free PMC article. Review.
-
Alpha kinase 3 signaling at the M-band maintains sarcomere integrity and proteostasis in striated muscle.Nat Cardiovasc Res. 2023 Feb;2(2):159-173. doi: 10.1038/s44161-023-00219-9. Epub 2023 Feb 15. Nat Cardiovasc Res. 2023. PMID: 39196058 Free PMC article.
-
Loss of epigenetic regulator TET2 and oncogenic KIT regulate myeloid cell transformation via PI3K pathway.JCI Insight. 2018 Feb 22;3(4):e94679. doi: 10.1172/jci.insight.94679. eCollection 2018 Feb 22. JCI Insight. 2018. PMID: 29467326 Free PMC article.
-
The Role of HECT-Type E3 Ligase in the Development of Cardiac Disease.Int J Mol Sci. 2021 Jun 4;22(11):6065. doi: 10.3390/ijms22116065. Int J Mol Sci. 2021. PMID: 34199773 Free PMC article. Review.
References
-
- Jessup M. & Brozena S. Heart failure. N Engl J Med 348, 2007–2018 (2003). - PubMed
-
- Johansen H., Strauss B., Arnold J. M., Moe G. & Liu P. On the rise: The current and projected future burden of congestive heart failure hospitalization in Canada. The Canadian journal of cardiology 19, 430–435 (2003). - PubMed
-
- Rubart M. & Field L. J. Cardiac regeneration: repopulating the heart. Annu Rev Physiol 68, 29–49 (2006). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
