

Mapping Biological Networks from Quantitative Data-Independent Acquisition Mass Spectrometry: Data to Knowledge Pipelines
- PMID: 28150249
- PMCID: PMC6844627
- DOI: 10.1007/978-1-4939-6783-4_19
Mapping Biological Networks from Quantitative Data-Independent Acquisition Mass Spectrometry: Data to Knowledge Pipelines
Abstract
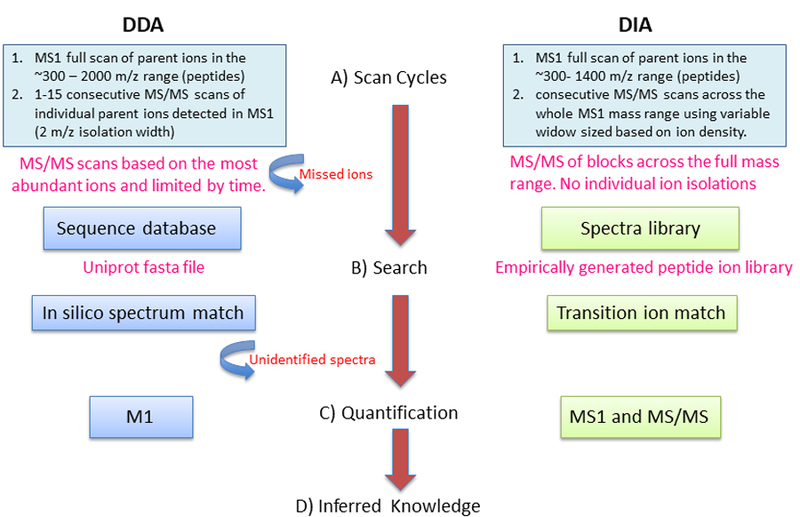
Data-independent acquisition mass spectrometry (DIA-MS) strategies and applications provide unique advantages for qualitative and quantitative proteome probing of a biological sample allowing constant sensitivity and reproducibility across large sample sets. These advantages in LC-MS/MS are being realized in fundamental research laboratories and for clinical research applications. However, the ability to translate high-throughput raw LC-MS/MS proteomic data into biological knowledge is a complex and difficult task requiring the use of many algorithms and tools for which there is no widely accepted standard and best practices are slowly being implemented. Today a single tool or approach inherently fails to capture the full interpretation that proteomics uniquely supplies, including the dynamics of quickly reversible chemically modified states of proteins, irreversible amino acid modifications, signaling truncation events, and, finally, determining the presence of protein from allele-specific transcripts. This chapter highlights key steps and publicly available algorithms required to translate DIA-MS data into knowledge.
Keywords: Citrullination; Data-independent acquisition; Phosphorylation; Post-translational modifications; Protein networks; SWATH.
Figures
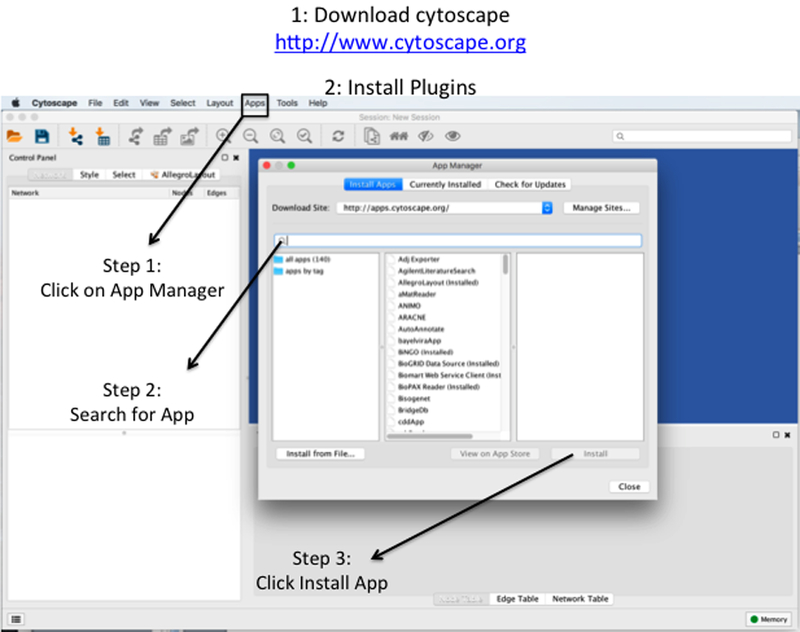
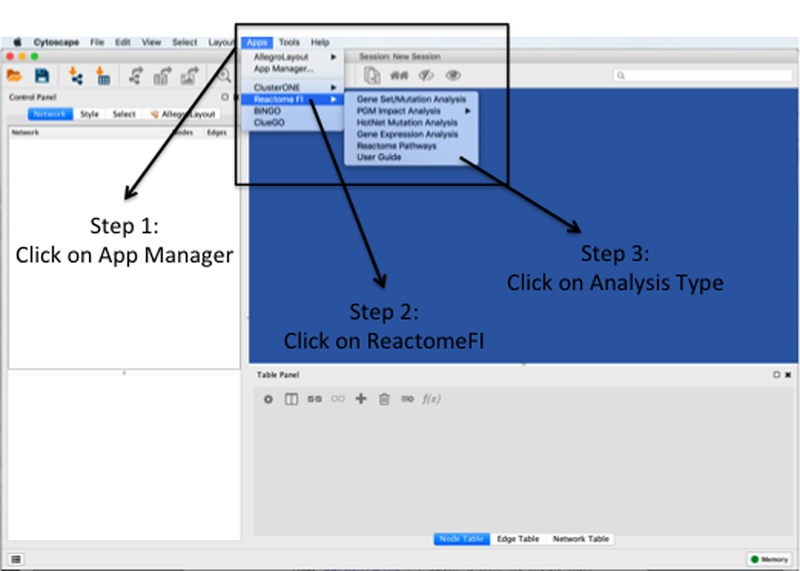
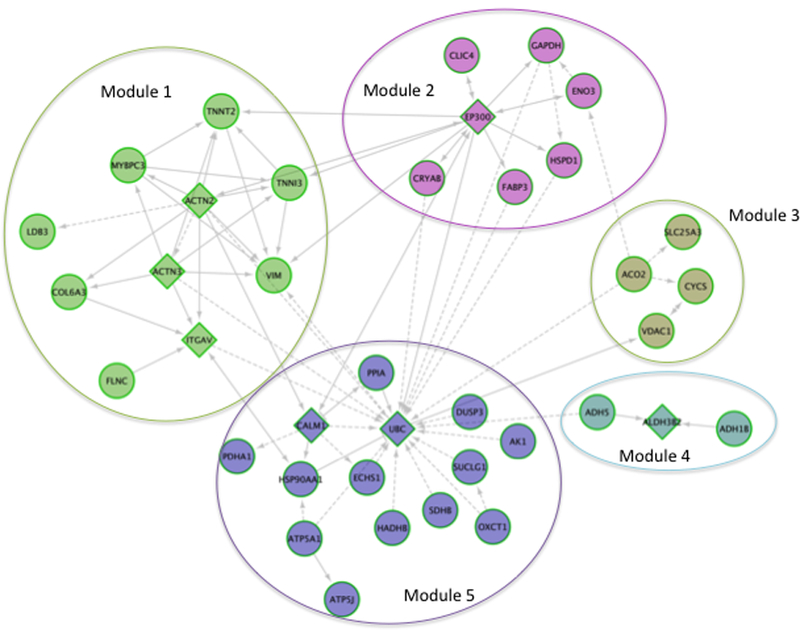
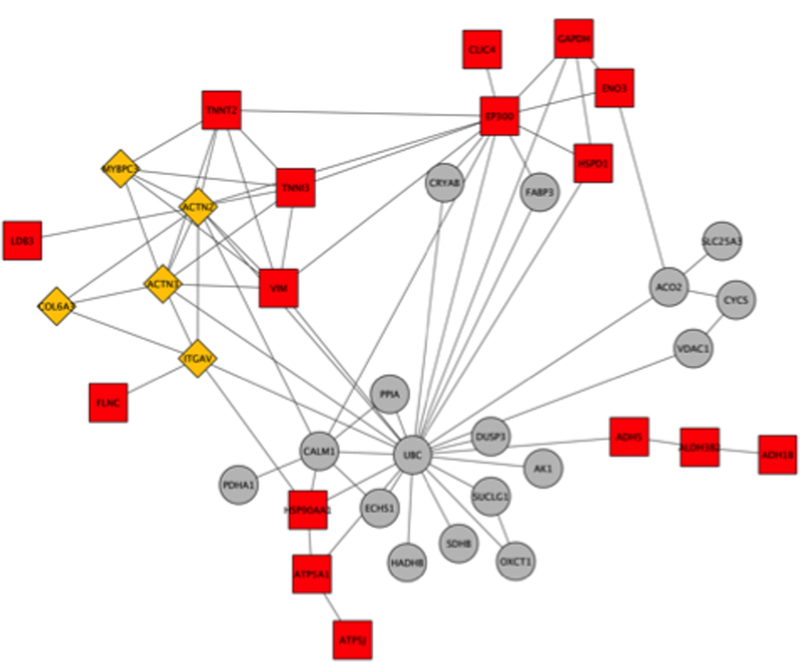
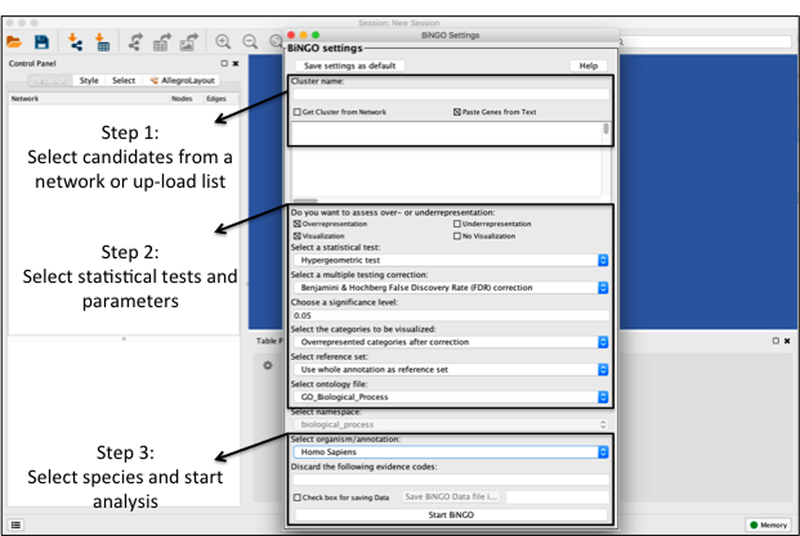
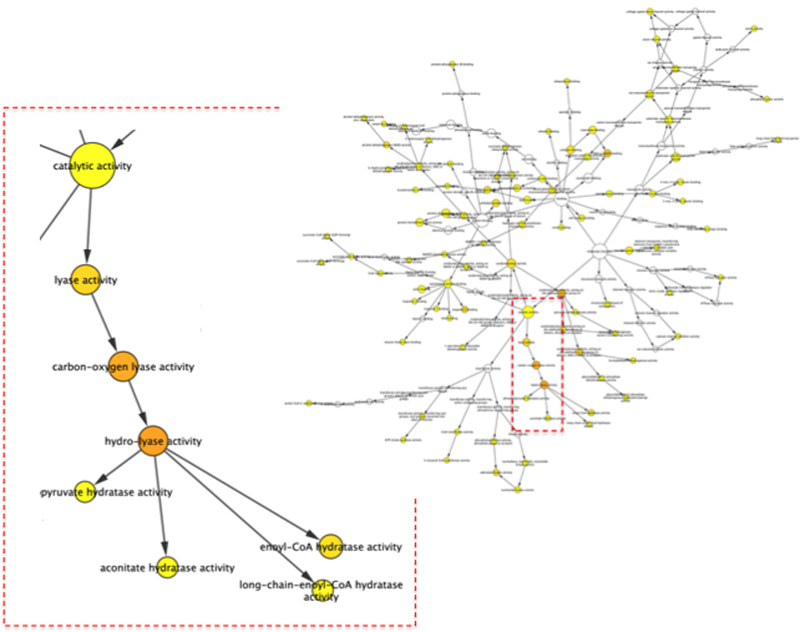
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
