

Mixture model normalization for non-targeted gas chromatography/mass spectrometry metabolomics data
- PMID: 28153035
- PMCID: PMC5290663
- DOI: 10.1186/s12859-017-1501-7
Mixture model normalization for non-targeted gas chromatography/mass spectrometry metabolomics data
Abstract
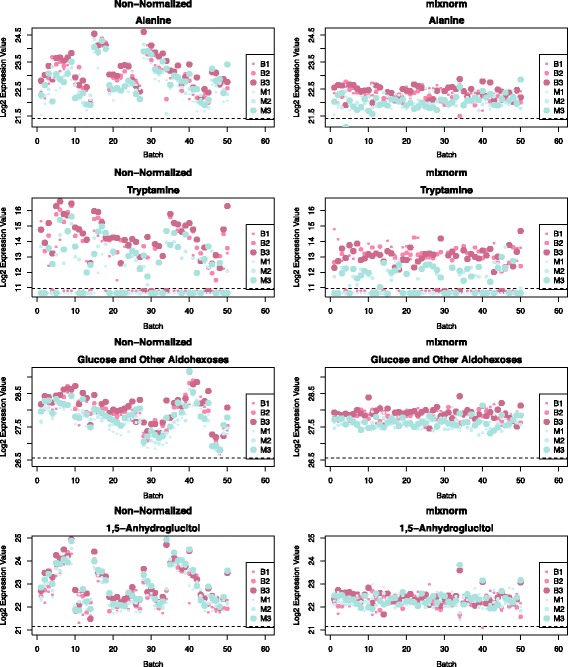
Background: Metabolomics offers a unique integrative perspective for health research, reflecting genetic and environmental contributions to disease-related phenotypes. Identifying robust associations in population-based or large-scale clinical studies demands large numbers of subjects and therefore sample batching for gas-chromatography/mass spectrometry (GC/MS) non-targeted assays. When run over weeks or months, technical noise due to batch and run-order threatens data interpretability. Application of existing normalization methods to metabolomics is challenged by unsatisfied modeling assumptions and, notably, failure to address batch-specific truncation of low abundance compounds.
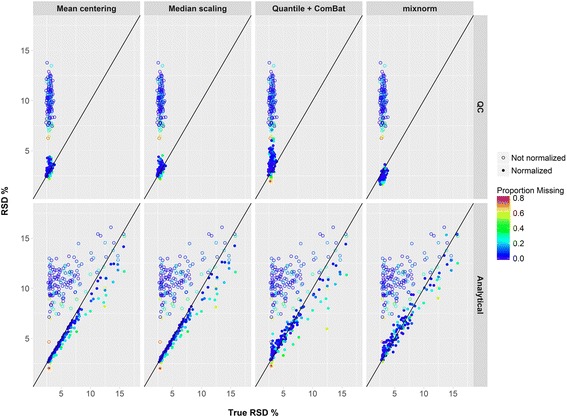
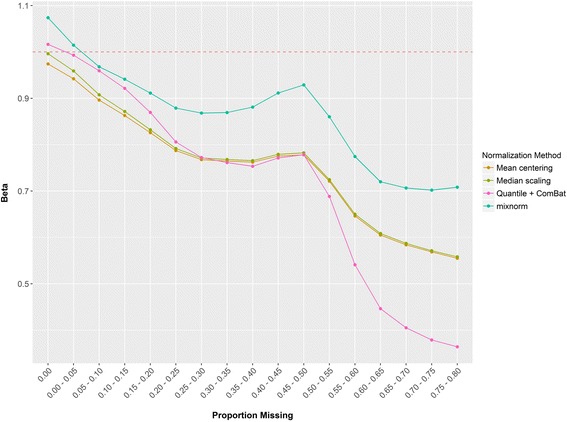
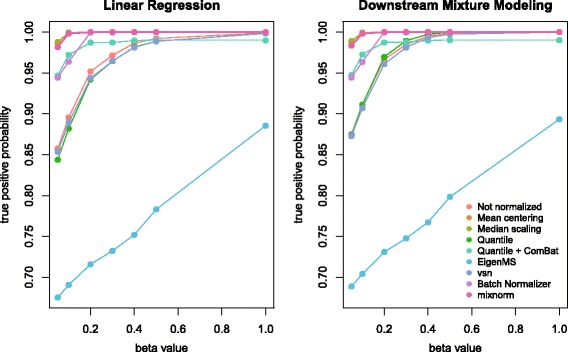
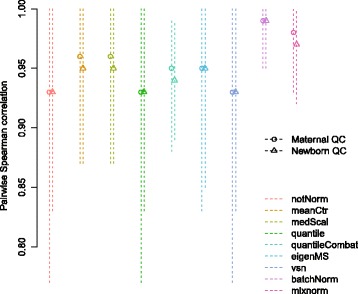
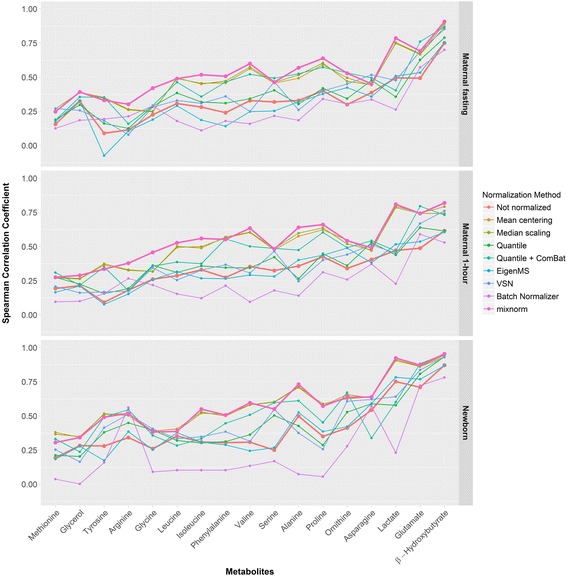
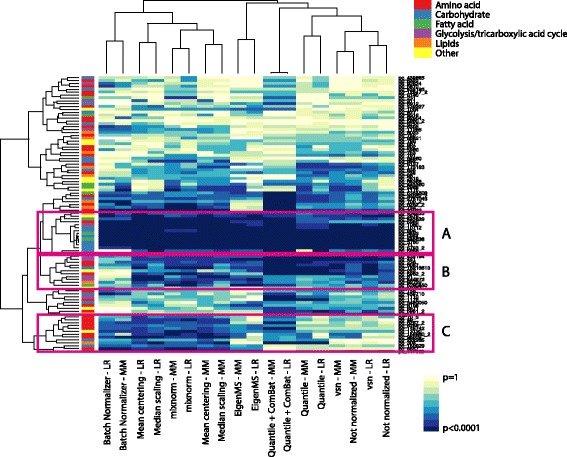
Results: To curtail technical noise and make GC/MS metabolomics data amenable to analyses describing biologically relevant variability, we propose mixture model normalization (mixnorm) that accommodates truncated data and estimates per-metabolite batch and run-order effects using quality control samples. Mixnorm outperforms other approaches across many metrics, including improved correlation of non-targeted and targeted measurements and superior performance when metabolite detectability varies according to batch. For some metrics, particularly when truncation is less frequent for a metabolite, mean centering and median scaling demonstrate comparable performance to mixnorm.
Conclusions: When quality control samples are systematically included in batches, mixnorm is uniquely suited to normalizing non-targeted GC/MS metabolomics data due to explicit accommodation of batch effects, run order and varying thresholds of detectability. Especially in large-scale studies, normalization is crucial for drawing accurate conclusions from non-targeted GC/MS metabolomics data.
Keywords: Batch effects; GC/MS; Gas chromatography/mass spectrometry; Metabolomics; Non-targeted; Normalization.
Figures

References
-
- Dunn WR, Broadhurst D, Begley P, Zelena E, Francis-McIntyre S, Anderson N, Brown M, Knowles JD, Halsall A, Haselden JN, et al. Procedures for large-scale metabolic profiling of serum and plasma using has chromatography and liquid chromatography coupled to mass spectrometry. Nat Protoc. 2011;6(7):1060–83. doi: 10.1038/nprot.2011.335. - DOI - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
