

CJD mimics and chameleons
- PMID: 28153848
- PMCID: PMC5520355
- DOI: 10.1136/practneurol-2016-001571
CJD mimics and chameleons
Abstract
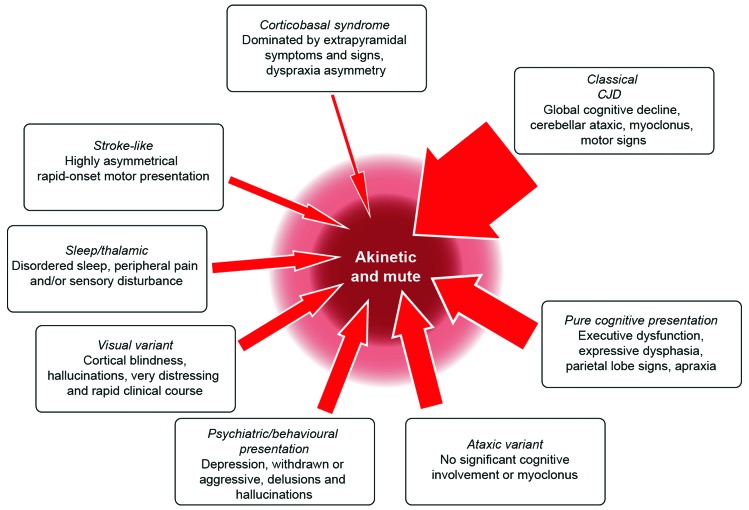
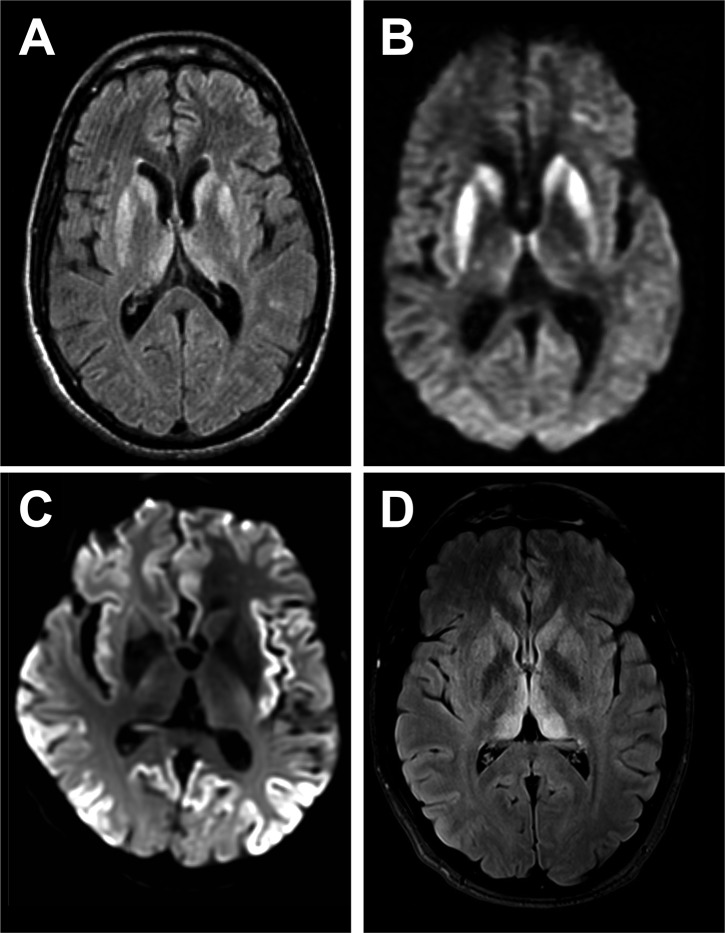
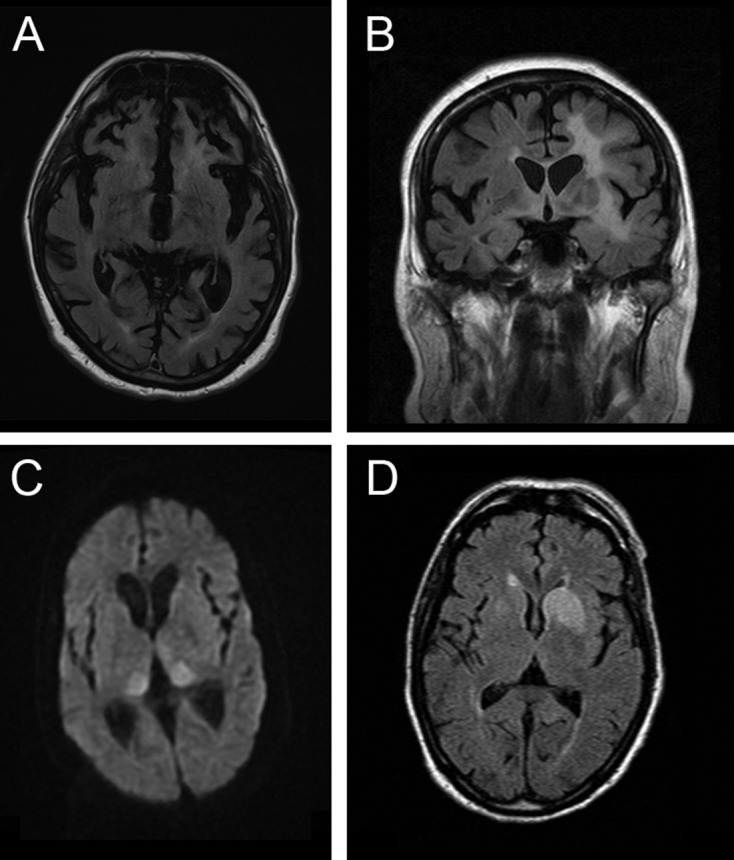
Rapidly progressive dementia mimicking Creutzfeldt-Jakob disease (CJD) is a relatively rare presentation but a rewarding one to become familiar with, as the potential diagnoses range from the universally fatal to the completely reversible. Patients require urgent decisions about assessment and investigation and have quickly evolving needs for treatments and support, through symptom management and end-of-life care in most cases. We have based this pragmatic review on the experiences of a specialist prion referral centre in the UK, which, unsurprisingly, is strongly biased towards seeing patients with CJD. Cases eventually proven not to have prion disease might be described as 'CJD-mimics'; being referred from UK neurologists, these are the most challenging cases. CJD in its classical presentation is very rarely mimicked; however, it is highly heterogeneous, and atypical forms can mimic virtually all common neurodegenerative syndromes. Warning features of a mimic include generalised seizures, hyponatraemia, fever, a facial movement disorder, a normal neurological examination and a modestly rapid presentation. Contrast-enhancing lesions or MRI signal hyperintensity outside the striatum, thalamus or cortex and a cerebrospinal fluid pleocytosis are key investigation pointers to a CJD mimic.
Keywords: rapidly progressive dementia.
© Article author(s) (or their employer(s) unless otherwise stated in the text of the article) 2017. All rights reserved. No commercial use is permitted unless otherwise expressly granted.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical